Ewing's sarcoma
| Ewing's sarcoma | |
|---|---|
| Other names | Ewing sarcoma, peripheral primitive neuroectodermal tumor, Askin tumor, and Ewing sarcoma family of tumors[1] |
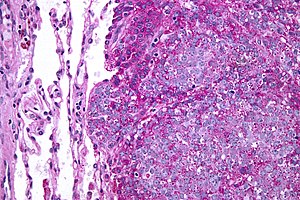 | |
| Micrograph of metastatic Ewing sarcoma (right of image) in normal lung (left of image). PAS stain. | |
| Pronunciation |
|
| Specialty | Oncology |
| Symptoms | Swell and pain near the tumor[1] |
| Complications | Pleural effusion, paraplegia[2] |
| Usual onset | 10 to 20 years old[3][2] |
| Causes | Unknown[2] |
| Diagnostic method | Tissue biopsy[1] |
| Differential diagnosis | Osteosarcoma, neuroblastoma, osteomyelitis, eosinophilic granuloma[2] |
| Treatment | Chemotherapy, radiation therapy, surgery, stem cell transplant[1] |
| Prognosis | Five year survival ~ 70%[3] |
| Frequency | 1 per million people (US)[3] |
Ewing sarcoma is a type of cancer that may be a bone sarcoma or a soft-tissue sarcoma.[1] Symptoms may include swelling and pain at the site of the tumor, fever, and a bone fracture.[1] The most common areas where it begins are the legs, pelvis, and chest wall.[3] In about 25% of cases, the cancer has already spread to other parts of the body at the time of diagnosis.[3] Complications may include a pleural effusion or paraplegia.[2]
The cause of Ewing sarcoma is unknown.[2] Most cases appear to occur randomly.[2] It is sometimes grouped together with primitive neuroectodermal tumors, in a category known as the Ewing family of tumors.[2] The underlying mechanism often involves a genetic change known as a reciprocal translocation.[2] Diagnosis is based on biopsy of the tumor.[1]
Treatment often includes chemotherapy, radiation therapy, surgery, and stem cell transplant.[1] Targeted therapy and immunotherapy are being studied.[1] Five year survival is about 70%.[3] A number of factors, however, affect this estimate.[3]
James Ewing in 1920 established that the tumor is a distinct type of cancer.[4][5] It affects about one in a million people per year in the United States.[3] Ewing sarcoma occurs most often in teenagers and young adults and represents 2% of childhood cancers.[1][2] Caucasians are affected more often than African Americans or Asians.[3] Males are affected more often than females.[3]
Signs and symptoms[]

Ewing sarcoma is more common in males (1.6 male:1 female) and usually presents in childhood or early adulthood, with a peak between 10 and 20 years of age. It can occur anywhere in the body but most commonly in the pelvis and proximal long tubular bones, especially around the growth plates. The diaphyses of the femur are the most common sites, followed by the tibia and the humerus. Thirty percent are overtly metastatic at presentation, while 10–15% of people present with a pathologic fracture at the time of diagnosis.[6] People usually experience extreme bone pain. Rarely, it can develop in the vagina.[7][8]
Signs and symptoms include intermittent fevers, anemia, leukocytosis, increased sedimentation rate, and other symptoms of inflammatory systemic illness.[9]
According to the Bone Cancer Research Trust (BCRT), the most common symptoms are localized pain, swelling, and sporadic bone pain with variable intensity. The swelling is most likely to be visible if the sarcoma is located on a bone near the surface of the body, but when it occurs in other places deeper in the body, like on the pelvis, it may not be visible.[10]
Genetics[]
Genetic exchange between chromosomes can cause cells to become cancerous. Most cases of Ewing sarcoma (about 85%) are the result of a defining genetic event; a reciprocal translocation between chromosomes 11 and 22, t(11,22), which fuses the Ewing's Sarcoma Breakpoint Region 1(EWSR1) gene of chromosome 22 (which encodes the EWS protein) to the Friend Leukemia Virus Integration 1 (FLI1) gene (which encodes Friend Leukemia Integration 1 transcription factor (FLI1), a member of the ETS transcription factor family) of chromosome 11.[9][6] The resultant chromosomal translocation causes the EWS trans-activation domain (which is usually silent in the wild type) to become very active, this leads to the translation of a new EWS-FLI1 fusion protein.[6] EWS proteins are involved in meiosis, B-lymphocyte maturation, hematopoietic stem cell renewal, DNA repair and cell senescence.[6] ETS transcription factors are involved in cell differentiation and cell cycle control.[6] The EWS-FLI1 fusion protein has phase transition properties, allowing it to transition into liquid-like, phase separated compartments consisting of membrane-less organelles. This phase transition property allows the fusion protein to access and activate micro-satellite regions of the genome that would otherwise be inaccessible. This fusion protein can convert usually silent chromatin regions into fully active enhancers leading to oncogenesis of the cells.[6]
The EWS-FLI1 fusion protein also causes variable expression of the genome via epigenetic mechanisms. The fusion protein does this by recruiting enzymes that affect DNA methylation, histone acetylation and direct inhibition of non-coding microRNA.[6] EWS-FLI1 promotes histone acetylation, which leads to uncoiling of DNA (which is usually tightly wound around histones); this chromatin relaxation leads to the DNA being more accessible to transcription factors and thus enhancing the expression of the associated genes.[6] DNA methylation leads to gene silencing as it prevents transcription factor binding. EWS-FLI1 reduces DNA methylation (which occurs mostly in areas corresponding to transcription enhancers), leading to increased gene expression.[6] The EWS-FLI1 fusion protein inhibits certain microRNAs of cells (such as miRNA-145). MiRNA-145 normally activates RNA-induced silencing complexes (RISCs) to inhibit or degrade mRNA that is involved in cell pluripotency.[6] Thus, ESW-FLI1 inhibition of the microRNA miRNA-145 leads to increased pluripotency and decreased differentiation of cells and increased oncogenesis.[6]
A genome-wide association study (GWAS) identified three susceptibility loci located on chromosomes 1, 10 and 15.[11] A continuative study discovered that the Ewing sarcoma susceptibility gene EGR2, which is located within the chromosome 10 susceptibility locus, is regulated by the EWSR1-FLI1 fusion oncogene via a GGAA-microsatellite.[12][13]
EWS/FLI functions as the master regulator.[14] Other translocations are at t(21;22)[15] and t(7;22).[16] Ewing sarcoma cells are positive for CD99 and MIC2,[9] and negative for CD45.[17]
Diagnosis[]

The definitive diagnosis is based on histomorphologic findings, immunohistochemistry and molecular pathology.
Ewing sarcoma is a small-blue-round-cell tumor that typically has a clear cytoplasm on H&E staining, due to glycogen. The presence of the glycogen can be demonstrated with positive PAS staining and negative PAS diastase staining. The characteristic immunostain is CD99, which diffusely marks the cell membrane. However, as CD99 is not specific for Ewing sarcoma, several auxiliary immunohistochemical markers can be employed to support the histological diagnosis.[18] Morphologic and immunohistochemical findings are corroborated with an associated chromosomal translocation, of which several occur. The most common translocation, present in about 90% of Ewing sarcoma cases, is t(11;22)(q24;q12),[19][20] which generates an aberrant transcription factor through fusion of the EWSR1 gene with the FLI1 gene.[21]
The pathologic differential diagnosis is the grouping of small-blue-round-cell tumors, which includes lymphoma, alveolar rhabdomyosarcoma, and desmoplastic small round cell tumor, among others.[citation needed]
Medical imaging[]
This section needs additional citations for verification. (February 2019) |

On conventional radiographs, typical findings of Ewing's Sarcoma consist of multiple confluent lytic bone lesions that have a "moth eaten" pattern due to permeative destruction of bone.[6] There will also be a displaced periosteum as the new sub-periosteal layer of bone begins to grow on top of the tumor. This raised or displaced periosteum is consistent with the classic radiographic finding of the Codman triangle.[6] The proliferative reaction of bone can also create delicate laminations constituting the periosteal layers and giving the radiographic appearance of an onion peel.[6] Plain films add valuable information in the initial evaluation or screening. The wide zone of transition (e.g. permeative) is the most useful plain film characteristic in differentiation of benign versus aggressive or malignant lytic lesions.

Magnetic resonance imaging (MRI) should be routinely used in the work-up of malignant tumors. It will show the full bony and soft tissue extent and relate the tumor to other nearby anatomic structures (e.g. vessels). Gadolinium contrast is not necessary as it does not give additional information over noncontrast studies, though some current researchers argue that dynamic, contrast-enhanced MRI may help determine the amount of necrosis within the tumor, thus help in determining response to treatment prior to surgery.[22]
Computed axial tomography (CT) can also be used to define the extraosseous extent of the tumor, especially in the skull, spine, ribs, and pelvis. Both CT and MRI can be used to follow response to radiation and/or chemotherapy. Bone scintigraphy can also be used to follow tumor response to therapy.[23]
In the group of malignant small round cell tumors that includes Ewing sarcoma, bone lymphoma, and small cell osteosarcoma, the cortex may appear almost normal radiographically, while permeative growth occurs throughout the Haversian channels. These tumors may be accompanied by a large soft-tissue mass while almost no bone destruction is visible. The radiographs frequently do not show any signs of cortical destruction.
Radiographically, Ewing sarcoma presents as "moth-eaten" destructive radiolucencies of the medulla and erosion of the cortex with expansion.[24]
Differential diagnosis[]
Other entities with similar clinical presentations include osteomyelitis, osteosarcoma (especially telangiectatic osteosarcoma), and eosinophilic granuloma. Soft-tissue neoplasms such as pleomorphic undifferentiated sarcoma (malignant fibrous histiocytoma) that erode into adjacent bone may also have a similar appearance. Accumulating evidence suggests that EWSR1-NFATc2 positive sarcomas, which were previously considered to possibly belong to the Ewing family of tumors, differ from Ewing sarcoma in their genetics, transcriptomes, and epigentic and epidemiological profiles, indicating that they might represent a distinct tumor entity.[25][26][27][28]
Treatment[]
Almost all people receive multidrug chemotherapy (most often vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide),[29] as well as local disease control with surgery and/or radiation.[30] An aggressive approach is necessary because almost all people with apparently localized disease at the time of diagnosis actually have asymptomatic metastatic disease.[31]
The surgical resection may involve limb salvage or amputation. Complete excision at the time of biopsy may be performed if malignancy is confirmed at the time it is examined.[citation needed] Treatment lengths vary depending on location and stage of the disease at diagnosis. Radical chemotherapy may be as short as six treatments at three-week cycles, but most people undergo chemotherapy for 6–12 months and radiation therapy for 5–8 weeks.[citation needed] Radiotherapy has been used for localized disease. The tumor has a unique property of being highly sensitive to radiation, sometimes acknowledged by the phrase "melting like snow", but the main drawback is that it recurs dramatically after some time.[citation needed]
Antisense oligodeoxynucleotides have been proposed as possible treatment by down-regulating the expression of the oncogenic fusion protein associated with the development of Ewing sarcoma resulting from the EWS-ETS gene translocation.[32][33] In addition, the synthetic retinoid derivative fenretinide (4-hydroxy(phenyl)retinamide) has been reported to induce high levels of cell death in Ewing sarcoma cell lines in vitro and to delay growth of xenografts in in vivo mouse models.[34][35]
In most pediatric cancers including sarcoma, proton beam radiation (also known as proton therapy) delivers an equally effective dose to the tumor with less damage to the surrounding normal tissue compared to photon radiation.[36]
Prognosis[]
Staging attempts to distinguish people with localized from those with metastatic disease.[37] The most common areas of metastasis are the lungs, bone and bone marrow with less common areas of metastasis being the lymph nodes, liver and brain.[6] The presence of metastatic disease is the most important prognostic factor in Ewing's Sarcoma with the 5 year survival rate being only 30% when metastasis is present at the time of diagnosis as compared to a 70% 5 year survival rate with no metastasis present.[6] Another important prognostic factor is the location of the primary tumor; proximal tumors (located in the pelvis and sacrum) are worse prognostic indicators as compared to more distal tumors.[6] Other factors associated with a poor prognosis include a large primary neoplasm, older age at diagnosis (older than 18 years of age) and increased lactate dehydrogenase (LDH) levels.[6]
Five-year survival for localized disease is greater than 70% after therapy.[38] Prior to the use of multi-drug chemotherapy, long-term survival was less than 10%. The development of multi-disciplinary therapy with chemotherapy, irradiation, and surgery has increased current long-term survival rates in most clinical centers to greater than 50%.[39] However, some sources state it is 25–30%.[40]
Retrospective research showed that two chemokine receptors, CXCR4 and CXCR7, can be used as molecular prognosis factors. People who express low levels of both chemokine receptors have the highest odds of long-term survival with >90% survival at five years post-diagnosis versus <30% survival at five years for patients with very high expression levels of both receptors.[41] A recent study also suggested a role for SOX2 as an independent prognostic biomarker that can be used to identify patients at high risk for tumor relapse.[42]
Epidemiology[]
Ewing sarcomas represent 16% of primary bone sarcomas.[9] In the United States, they are most common in the second decade of life,[9] with a rate of 0.3 cases per million in children under 3 years of age, and as high as 4.6 cases per million in adolescents aged 15–19 years. Internationally, the annual incidence rate averages less than 2 cases per million children.[43]
In the United Kingdom, an average of six children per year is diagnosed, mainly males in early stages of puberty. Due to the prevalence of diagnosis during teenage years, a link may exist between the onset of puberty and the early stages of this disease, although no research confirms this hypothesis.[citation needed]
A grouping of three unrelated teenagers in Wake Forest, North Carolina, have been diagnosed with Ewing sarcoma. All three children were diagnosed in 2011 and all attended the same temporary classroom together while the school underwent renovation. A fourth teenager living nearby was diagnosed in 2009. The odds of this grouping are considered significant.[44] Ewing sarcoma occurs about 10- to 20-fold more commonly in people of European descent compared to people of African descent.[45][6]
Ewing sarcoma is the second most common bone cancer in children and adolescents, with poor prognosis and outcome in ~70% of initial diagnoses and 10–15% of relapses.[46]
References[]
- ^ Jump up to: a b c d e f g h i j "Ewing Sarcoma Treatment". National Cancer Institute. 25 January 2019. Retrieved 3 February 2019.
- ^ Jump up to: a b c d e f g h i j "Ewing Sarcoma". NORD (National Organization for Rare Disorders). 2013. Retrieved 4 February 2019.
- ^ Jump up to: a b c d e f g h i j "Ewing Sarcoma Treatment". National Cancer Institute. 31 January 2019. Retrieved 4 February 2019.
- ^ "Ewing's sarcoma". Whonamedit. Retrieved 4 February 2019.
- ^ Ewing J (September 2006). "The Classic: Diffuse endothelioma of bone. Proceedings of the New York Pathological Society. 1921;12:17". Clinical Orthopaedics and Related Research. 450: 25–7. doi:10.1097/01.blo.0000229311.36007.c7. PMID 16951641.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s Riggi, Nicolò; Suvà, Mario L.; Stamenkovic, Ivan (14 January 2021). "Ewing's Sarcoma". New England Journal of Medicine. 384 (2): 154–164. doi:10.1056/NEJMra2028910. PMID 33497548. S2CID 231762844.
- ^ "Tumours of the Vagina; Chapter Six" (PDF). International Agency for Research on Cancer, World Health Organization. pp. 291–311. Archived from the original (PDF) on 2015-09-08. Retrieved 2018-03-14.
- ^ "Vulva and Vagina tumors: an overview". Atlas of Genetics and Cytogenetics in Oncology and Haematology. Archived from the original on 2018-02-22. Retrieved 2018-03-14.
- ^ Jump up to: a b c d e Goldman L, Cecil RL, Schafer AI (2012). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 1326. ISBN 978-1-4377-2788-3. OCLC 909785616.
- ^ "Symptoms of Ewing's Sarcoma". Bone Cancer Research Trust. October 2010. Archived from the original on 2013-01-30. Retrieved 2012-11-05.
- ^ Postel-Vinay S, Véron AS, Tirode F, Pierron G, Reynaud S, Kovar H, et al. (February 2012). "Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma". Nature Genetics. 44 (3): 323–7. doi:10.1038/ng.1085. PMID 22327514. S2CID 205343425.
- ^ Grünewald TG, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, Aynaud MM, et al. (September 2015). "Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite". Nature Genetics. 47 (9): 1073–8. doi:10.1038/ng.3363. PMC 4591073. PMID 26214589.
- ^ Gomez NC, Davis IJ (September 2015). "Linking germline and somatic variation in Ewing sarcoma". Nature Genetics. 47 (9): 964–5. doi:10.1038/ng.3387. PMID 26313223. S2CID 5454861.
- ^ Owen LA, Kowalewski AA, Lessnick SL (April 2008). "EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing sarcoma". PLOS ONE. 3 (4): e1965. Bibcode:2008PLoSO...3.1965O. doi:10.1371/journal.pone.0001965. PMC 2291578. PMID 18414662.
- ^ Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT (February 1994). "A second Ewing sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG". Nature Genetics. 6 (2): 146–51. doi:10.1038/ng0294-146. PMID 8162068. S2CID 19747268.
- ^ Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, Shapiro DN (March 1995). "A variant Ewing's sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1". Oncogene. 10 (6): 1229–34. PMID 7700648.
- ^ Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, Juergens H (May 2006). "Ewing's sarcoma family of tumors: current management". The Oncologist. 11 (5): 503–19. doi:10.1634/theoncologist.11-5-503. PMID 16720851.
- ^ McCuiston A, Bishop JA (March 2018). "Usefulness of NKX2.2 Immunohistochemistry for Distinguishing Ewing Sarcoma from Other Sinonasal Small Round Blue Cell Tumors". Head and Neck Pathology. 12 (1): 89–94. doi:10.1007/s12105-017-0830-1. PMC 5873485. PMID 28616785.
- ^ "Soft tissue tumors: Ewing's tumors/Primitive neurectodermal tumors (PNET)". Atlas of Genetics and Cytogenetics in Oncology and Haematology. Archived from the original on 29 October 2012. Retrieved 5 November 2012.
- ^ Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C, Thiery JP, Olschwang S, Philip I, Berger MP (June 1988). "Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12)". Cancer Genetics and Cytogenetics. 32 (2): 229–38. doi:10.1016/0165-4608(88)90285-3. PMID 3163261.
- ^ Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G (September 1992). "Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours". Nature. 359 (6391): 162–5. Bibcode:1992Natur.359..162D. doi:10.1038/359162a0. PMID 1522903. S2CID 4331584.
- ^ "Ewing Sarcoma". The Lecturio Medical Concept Library. Retrieved 22 July 2021.
- ^ "Ewing Sarcoma". The Lecturio Medical Concept Library. Retrieved 22 July 2021.
- ^ "Ewing Sarcoma". The Lecturio Medical Concept Library. Retrieved 22 July 2021.
- ^ Grünewald TG, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H, et al. (July 2018). "Ewing sarcoma". Nature Reviews. Disease Primers. 4 (1): 5. doi:10.1038/s41572-018-0003-x. PMID 29977059. S2CID 49571421.
- ^ Koelsche C, Hartmann W, Schrimpf D, Stichel D, Jabar S, Ranft A, et al. (August 2018). "Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information". Modern Pathology. 31 (8): 1246–1256. doi:10.1038/s41379-018-0045-3. PMC 7484949. PMID 29572501.
- ^ Baldauf MC, Gerke JS, Orth MF, Dallmayer M, Baumhoer D, de Alava E, et al. (June 2018). "Are EWSR1-NFATc2-positive sarcomas really Ewing sarcomas?". Modern Pathology. 31 (6): 997–999. doi:10.1038/s41379-018-0009-7. PMID 29895896.
- ^ Watson S, Perrin V, Guillemot D, Reynaud S, Coindre JM, Karanian M, et al. (May 2018). "Transcriptomic definition of molecular subgroups of small round cell sarcomas". The Journal of Pathology. 245 (1): 29–40. doi:10.1002/path.5053. PMID 29431183.
- ^ Lahl M, Fisher VL, Laschinger K (February 2008). "Ewing's sarcoma family of tumors: an overview from diagnosis to survivorship". Clinical Journal of Oncology Nursing. 12 (1): 89–97. doi:10.1188/08.CJON.89-97. PMID 18258578. S2CID 10512706.
- ^ Randall L, Calvert G, Spraker H, Lessnick S (2005). "Ewing's Sarcoma Family of Tumours (ESFT)". Liddy Shriver Sarcoma Initiative. Archived from the original on 2009-02-08. Retrieved 2009-04-15.
- ^ Salazar, Luis; Ghali, Abdullah. "An Ankle Sprain or Calcaneal Ewing Sarcoma A case report of a 9 year old boy". Biomedical Research and Clinical Reviews. 3.
- ^ Asami S, Chin M, Shichino H, Yoshida Y, Nemoto N, Mugishima H, Suzuki T (March 2008). "Treatment of Ewing's sarcoma using an antisense oligodeoxynucleotide to regulate the cell cycle". Biological & Pharmaceutical Bulletin. 31 (3): 391–4. doi:10.1248/bpb.31.391. PMID 18310898.
- ^ Mateo-Lozano S, Gokhale PC, Soldatenkov VA, Dritschilo A, Tirado OM, Notario V (November 2006). "Combined transcriptional and translational targeting of EWS/FLI-1 in Ewing's sarcoma". Clinical Cancer Research. 12 (22): 6781–90. doi:10.1158/1078-0432.CCR-06-0609. PMID 17121899. S2CID 1404471.
- ^ Myatt SS, Redfern CP, Burchill SA (April 2005). "p38MAPK-Dependent sensitivity of Ewing's sarcoma family of tumors to fenretinide-induced cell death". Clinical Cancer Research. 11 (8): 3136–48. doi:10.1158/1078-0432.CCR-04-2050. PMID 15837770.
- ^ Myatt SS, Burchill SA (February 2008). "The sensitivity of the Ewing's sarcoma family of tumours to fenretinide-induced cell death is increased by EWS-Fli1-dependent modulation of p38(MAPK) activity". Oncogene. 27 (7): 985–96. doi:10.1038/sj.onc.1210705. PMID 17700534.
- ^ Ladra MM, Yock TI (January 2014). "Proton radiotherapy for pediatric sarcoma". Cancers. 6 (1): 112–27. doi:10.3390/cancers6010112. PMC 3980591. PMID 24424260.
- ^ McTiernan AM, Cassoni AM, Driver D, Michelagnoli MP, Kilby AM, Whelan JS (2006). "Improving Outcomes After Relapse in Ewing's Sarcoma: Analysis of 114 Patients From a Single Institution". Sarcoma. 2006: 1–8. doi:10.1155/SRCM/2006/83548. PMC 1698143. PMID 17496997.
- ^ "How Is the Ewing Family of Tumors Staged?". American Cancer Society. 19 June 2006. Archived from the original on 2008-04-22.
- ^ Iwamoto Y (February 2007). "Diagnosis and treatment of Ewing's sarcoma". Japanese Journal of Clinical Oncology. 37 (2): 79–89. doi:10.1093/jjco/hyl142. PMID 17272319.
- ^ Thacker MM, Temple HT, Scully SP (April 2005). "Current treatment for Ewing's sarcoma". Expert Review of Anticancer Therapy. 5 (2): 319–31. doi:10.1586/14737140.5.2.319. PMID 15877528. S2CID 26773908.
- ^ Bennani-Baiti IM, Cooper A, Lawlor ER, Kauer M, Ban J, Aryee DN, Kovar H (July 2010). "Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing's sarcoma". Clinical Cancer Research. 16 (14): 3769–78. doi:10.1158/1078-0432.CCR-10-0558. PMC 2905506. PMID 20525755.
- ^ Sannino G, Marchetto A, Ranft A, Jabar S, Zacherl C, Alba-Rubio R, Stein S, Wehweck FS, Kiran MM, Hoelting TL, Musa J (2018-12-17). "SOX2 expression identifies Ewing sarcoma patients with high risk for tumor relapse and poor survival". bioRxiv: 498253. doi:10.1101/498253.
- ^ Ewing Sarcoma Imaging at eMedicine
- ^ "Three Wake students battle rare cancer: Cluster or coincidence?". WRAL.com. 29 April 2013. Archived from the original on 2013-05-01. Retrieved 2013-04-30.
- ^ Worch J, Cyrus J, Goldsby R, Matthay KK, Neuhaus J, DuBois SG (March 2011). "Racial differences in the incidence of mesenchymal tumors associated with EWSR1 translocation". Cancer Epidemiology, Biomarkers & Prevention. 20 (3): 449–53. doi:10.1158/1055-9965.EPI-10-1170. PMC 3051020. PMID 21212061.
- ^ Twardziok M, Kleinsimon S, Rolff J, Jäger S, Eggert A, Seifert G, Delebinski CI (2016). "Multiple Active Compounds from Viscum album L. Synergistically Converge to Promote Apoptosis in Ewing Sarcoma". PLOS ONE. 11 (9): e0159749. Bibcode:2016PLoSO..1159749T. doi:10.1371/journal.pone.0159749. PMC 5010293. PMID 27589063.
Further reading[]
- "Ewing family of tumors". NCI Dictionary of Cancer Terms. 2011-02-02.
- van der Woude HJ, Smithuis R (2010-04-10). "Differential diagnosis of bone tumors". Leiderdorp, the Netherlands: Radiology department of the Onze Lieve Vrouwe Gasthuis, Amsterdam and the Rijnland hospital.
| Classification | |
|---|---|
| External resources |
- Osseous and chondromatous neoplasia
- Pediatric cancers
- Rare diseases
- Sarcoma
- Small-blue-round-cell tumors