High Resolution Melt
This article has multiple issues. Please help or discuss these issues on the talk page. (Learn how and when to remove these template messages)
|
High Resolution Melt (HRM) analysis is a powerful technique in molecular biology for the detection of mutations, polymorphisms and epigenetic differences in double-stranded DNA samples. It was discovered and developed by Idaho Technology and the University of Utah.[1] It has advantages over other genotyping technologies, namely:
- It is cost-effective vs. other genotyping technologies such as sequencing and TaqMan SNP typing. This makes it ideal for large scale genotyping projects.
- It is fast and powerful thus able to accurately genotype many samples rapidly.
- It is simple. With a good quality HRM assay, powerful genotyping can be performed by non-geneticists in any laboratory with access to an HRM capable real-time PCR machine.
Method[]
HRM analysis is performed on double stranded DNA samples. Typically the user will use polymerase chain reaction (PCR) prior to HRM analysis to amplify the DNA region in which their mutation of interest lies. In the sample tube there are now many copies of the DNA region of interest. This region that is amplified is known as the amplicon. After the PCR process the HRM analysis begins. The process is simply a precise warming of the amplicon DNA from around 50 ˚C up to around 95 ˚C. At some point during this process, the melting temperature of the amplicon is reached and the two strands of DNA separate or "melt" apart.

The key to HRM is to monitor this separation of strands in real-time. This is achieved by using a fluorescent dye. The dyes that are used for HRM are known as intercalating dyes and have a unique property. They bind specifically to double-stranded DNA and when they are bound they fluoresce brightly. In the absence of double stranded DNA they have nothing to bind to and they only fluoresce at a low level. At the beginning of the HRM analysis there is a high level of fluorescence in the sample because of the billions of copies of the amplicon. But as the sample is heated up and the two strands of the DNA melt apart, presence of double stranded DNA decreases and thus fluorescence is reduced. The HRM machine has a camera that watches this process by measuring the fluorescence. The machine then simply plots this data as a graph known as a melt curve, showing the level of fluorescence vs the temperature:

Comparison of melt curves[]
The melting temperature of the amplicon at which the two DNA strands come apart is entirely predictable. It is dependent on the sequence of the DNA bases. If you are comparing two samples from two different people, they should give exactly the same shaped melt curve. However, if one person has a mutation in the DNA region you have amplified, then this will alter the temperature at which the DNA strands melt apart. So now the two melt curves appear different. The difference may only be tiny, perhaps a fraction of a degree, but because the HRM machine has the ability to monitor this process in "high resolution", it is possible to accurately document these changes and therefore identify if a mutation is present or not.
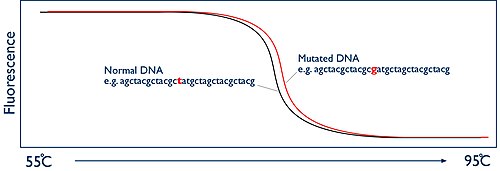
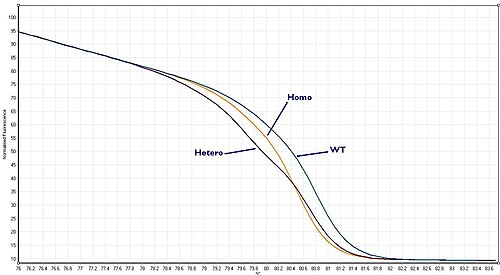
Wild type, heterozygote or homozygote?[]
Things become slightly more complicated than this because organisms contain two (or more) copies of each gene, known as the two alleles. So, if a sample is taken from a patient and amplified using PCR both copies of the region of DNA (alleles) of interest are amplified. So if we are looking for mutation there are now three possibilities:
- Neither allele contains a mutation
- One or other allele contains a mutation
- Both alleles contain a mutation.
These three scenarios are known as "Wild–type", "Heterozygote" or "Homozygote" respectively. Each gives a melt curve that is slightly different. With a high quality HRM assay it is possible to distinguish between all three of these scenarios.
Homozygous allelic variants may be characterised by a temperature shift on the resulting melt curve produced by HRM analysis. In comparison, heterozygotes are characterised by changes in melt curve shape. This is due to base-pair mismatching generated as a result of destabilised heteroduplex annealing between wild-type and variant strands. These differences can be easily seen on the resulting melt curve and the melt profile differences between the different genotypes can be amplified visually via generating a difference curve [2]
Applications[]
SNP typing/Point mutation detection[]
Conventional SNP typing methods are typically time-consuming and expensive, requiring several probe based assays to be multiplexed together or the use of DNA microarrays. HRM is more cost-effective and reduces the need to design multiple pairs of primers and the need to purchase expensive probes. The HRM method has been successfully used to detect a single G to A substitution in the gene Vssc (Voltage Sensitive Sodium Channel) which confers resistance to the acaricide permethrin in Scabies mite. This mutation results in a coding change in the protein (G1535D). The analysis of scabies mites collected from suspected permethrin susceptible and tolerant populations by HRM showed distinct melting profiles. The amplicons from the sensitive mites were observed to have a higher melting temperature relative to the tolerant mites, as expected from the higher thermostability of the GC base pair[3]
In a field more relevant to clinical diagnostics, HRM has been shown to be suitable in principle for the detection of mutations in the breast cancer susceptibility genes BRCA1 and BRCA2. More than 400 mutations have been identified in these genes.
The sequencing of genes is the gold standard for identifying mutations. Sequencing is time-consuming and labour-intensive and is often preceded by techniques used to identify heteroduplex DNA, which then further amplify these issues. HRM offers a faster and more convenient closed-tube method of assessing the presence of mutations and gives a result which can be further investigated if it is of interest. In a study carried out by Scott et al. in 2006,[4] 3 cell lines harbouring different BRCA mutations were used to assess the HRM methodology. It was found that the melting profiles of the resulting PCR products could be used to distinguish the presence or absence of a mutation in the amplicon. Similarly in 2007 Krypuy et al.[5] showed that the careful design of HRM assays (with regards to primer placement) could be successfully employed to detect mutations in the TP53 gene, which encodes the tumour suppressor protein p53 in clinical samples of breast and ovarian cancer. Both these studies highlighted the fact that changes in the melting profile can be in the form of a shift in the melting temperature or an obvious difference in the shape of the melt curve. Both of these parameters are a function of the amplicon sequence.
The consensus is that HRM is a cost efficient method that can be employed as an initial screen for samples suspected of harbouring polymorphisms or mutations. This would reduce the number of samples which need to be investigated further using more conventional methods.
Zygosity testing[]
Currently there are many methods used to determine the zygosity status of a gene at a particular locus. These methods include the use of PCR with specifically designed probes to detect the variants of the genes (SNP typing is the simplest case). In cases where longer stretches of variation is implicated, post PCR analysis of the amplicons may be required. Changes in enzyme restriction, electrophoretic and chromatographic profiles can be measured. These methods are usually more time-consuming and increase the risk of amplicon contamination in the laboratory, due to the need to work with high concentrations of amplicons in the lab post-PCR. The use of HRM reduces the time required for analysis and the risk of contamination. HRM is a more cost-effective solution and the high resolution element not only allows the determination of homo and heterozygosity, it also resolves information about the type of homo and heterozygosity, with different gene variants giving rise to differing melt curve shapes. A study by Gundry et al. 2003,[6] showed that fluorescent labelling of one primer (in the pair) has been shown to be favourable over using an intercalating dye such as SYBR green I. However, progress has been made in the development and use of improved intercalating dyes [7] which reduce the issue of PCR inhibition and concerns over non-saturating intercalation of the dye.
Epigenetics[]
The HRM methodology has also been exploited to provide a reliable analysis of the methylation status of DNA. This is of significance since changes to the methylation status of tumour suppressor genes, genes that regulate apoptosis and DNA repair, are characteristics of cancers and also have implications for responses to chemotherapy. For example, cancer patients can be more sensitive to treatment with DNA alkylating agents if the promoter of the DNA repair gene MGMT of the patient is methylated. In a study which tested the methylation status of the MGMT promoter on 19 colorectal samples, 8 samples were found to be methylated.[8] Another study compared the predictive power of MGMT promoter methylation in 83 high grade glioma patients obtained by either MSP, pyrosequencing, and HRM. The HRM method was found to be at least equivalent to pyrosequencing in quantifying the methylation level.[9]
Methylated DNA can be treated by bi-sulphite modification, which converts non-methylated cytosines to uracil. Therefore, PCR products resulting from a template that was originally unmethylated will have a lower melting point than those derived from a methylated template. HRM also offers the possibility of determining the proportion of methylation in a given sample, by comparing it to a standard curve which is generated by mixing different ratios of methylated and non-methylated DNA together. This can offer information regarding the degree of methylation that a tumour may have and thus give an indication of the character of the tumour and how far it deviates from what is "normal".
HRM also is practically advantageous for use in diagnostics, due to its capacity to be adapted to high throughput screening testing, and again it minimises the possibility of amplicon spread and contamination within a laboratory, owing to its closed-tube format.
Intercalating dyes[]
To follow the transition of dsDNA (double-stranded) to ssDNA (single-stranded), intercalating dyes are employed. These dyes show differential fluorescence emission dependent on their association with double-stranded or single-stranded DNA. SYBR Green I is a first generation dye for HRM. It fluoresces when intercalated into dsDNA and not ssDNA. Because it may inhibit PCR at high concentrations, it is used at sub-saturating concentrations. Recently, some researchers have discouraged the use of SYBR Green I for HRM,[10] claiming that substantial protocol modifications are required. This is because it is suggested that the lack of accuracy may result from "dye jumping", where dye from a melted duplex may get reincorporated into regions of dsDNA which had not yet melted.[6][10] New saturating dyes such as LC Green and LC Green Plus, ResoLight, EvaGreen, Chromofy and SYTO 9 are available on the market and have been used successfully for HRM. However, some groups have successfully used SYBR Green I for HRM with the Corbett Rotorgene instruments [11] and advocate the use of SYBR Green I for HRM applications.
Design of high-resolution melting experiments[]
High resolution melting assays typically involve qPCR amplification followed by a melting curve collected using a fluorescent dye. Due to the sensitivity of high-resolution melting analysis, it is necessary to carefully consider PCR cycling conditions, template DNA quality, and melting curve parameters.[12] For accurate and repeatable results, PCR thermal cycling conditions must be optimized to ensure that the desired DNA region is amplified with high specificity and minimal bias between sequence variants. The melting curve is typically performed across a broad range of temperatures in small (~0.3 °C) increments that are long enough (~10 seconds) for the DNA to reach equilibrium at each temperature step.
In addition to typical primer design considerations, the design of primers for high-resolution melting assays involves maximizing the thermodynamic differences between PCR products belonging to different genotypes. Smaller amplicons generally yield greater melting temperature variation than longer amplicons, but the variability cannot be predicted by eye. For this reason, it is critical to accurately predict the melting curve of PCR products when designing primers that will distinguish sequence variants. Specialty software, such as uMelt[13] and DesignSignatures,[14] are available to help design primers that will maximize melting curve variability specifically for high-resolution melting assays.
See also[]
- Beacon Designer
- Melting curve analysis
References[]
- ^ For academic treatment of the history of HRM see http://www.dna.utah.edu/Hi-Res/TOP_Hi-Res%20Melting.html
- ^ S Taylor et al., 2010. A Practical Guide To High Resolution Melt Analysis Genotyping. BioRad Tech Note 6004.
- ^ Pasay C, Arlian L, Morgan M, et al. (March 2008). "High-resolution melt analysis for the detection of a mutation associated with permethrin resistance in a population of scabies mites". Med. Vet. Entomol. 22 (1): 82–8. doi:10.1111/j.1365-2915.2008.00716.x. PMID 18380658.
- ^ James PA, Doherty R, Harris M, et al. (February 2006). "Optimal selection of individuals for BRCA mutation testing: a comparison of available methods". J. Clin. Oncol. 24 (4): 707–15. doi:10.1200/JCO.2005.01.9737. PMID 16446345.
- ^ Krypuy M, Ahmed AA, Etemadmoghadam D, et al. (2007). "High resolution melting for mutation scanning of TP53 exons 5-8". BMC Cancer. 7: 168. doi:10.1186/1471-2407-7-168. PMC 2025602. PMID 17764544.
- ^ a b Gundry CN, Vandersteen JG, Reed GH, Pryor RJ, Chen J, Wittwer CT (March 2003). "Amplicon melting analysis with labeled primers: a closed-tube method for differentiating homozygotes and heterozygotes". Clin. Chem. 49 (3): 396–406. doi:10.1373/49.3.396. PMID 12600951.
- ^ Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ (June 2003). "High-resolution genotyping by amplicon melting analysis using LCGreen". Clin. Chem. 49 (6 Pt 1): 853–60. doi:10.1373/49.6.853. PMID 12765979.
- ^ Wojdacz TK, Dobrovic A (2007). "Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation". Nucleic Acids Res. 35 (6): e41. doi:10.1093/nar/gkm013. PMC 1874596. PMID 17289753.
- ^ Switzeny, Olivier J.; Christmann, Markus; Renovanz, Mirjam; Giese, Alf; Sommer, Clemens; Kaina, Bernd (2016-05-05). "MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response". Clinical Epigenetics. 8: 49. doi:10.1186/s13148-016-0204-7. ISSN 1868-7083. PMC 4858829. PMID 27158275.
- ^ a b Reed GH, Kent JO, Wittwer CT (June 2007). "High-resolution DNA melting analysis for simple and efficient molecular diagnostics". Pharmacogenomics. 8 (6): 597–608. doi:10.2217/14622416.8.6.597. PMID 17559349. as PDF
- ^ Pornprasert S, Phusua A, Suanta S, Saetung R, Sanguansermsri T (June 2008). "Detection of alpha-thalassemia-1 Southeast Asian type using real-time gap-PCR with SYBR Green1 and high resolution melting analysis". Eur. J. Haematol. 80 (6): 510–4. CiteSeerX 10.1.1.509.2403. doi:10.1111/j.1600-0609.2008.01055.x. PMID 18284625.
- ^ Montgomery JL, Sanford LN, Wittwer CT (2010). "High-resolution DNA melting analysis in clinical research and diagnostics". Expert Rev Mol Diagn. 10 (2): 219–240. doi:10.1586/erm.09.84. PMID 20214540.
- ^ Dwight Z, Palais R, Wittwer CT (2011). "uMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application". Bioinformatics. 27 (7): 1019–1020. doi:10.1093/bioinformatics/btr065. PMID 21300699.
- ^ Wright ES, Vetsigian KH (2016). "DesignSignatures: a tool for designing primers that yields amplicons with distinct signatures". Bioinformatics. 32 (10): 1565–1567. doi:10.1093/bioinformatics/btw047. PMID 26803162.
External links[]
- Molecular biology
- DNA
- Biotechnology