Presynaptic inhibition
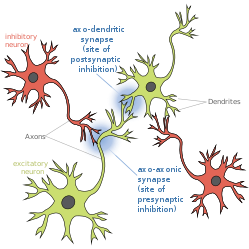
Presynaptic inhibition is a phenomenon in which an inhibitory neuron provides synaptic input to the axon of another neuron (axo-axonal synapse) to make it less likely to fire an action potential. Presynaptic inhibition occurs when an inhibitory neurotransmitter, like GABA, acts on GABA receptors on the axon terminal. Presynaptic inhibition is ubiquitous among sensory neurons.[1]
Function of presynaptic inhibition[]
Somatosensory neurons encode information about the body's current state (e.g. temperature, pain, pressure, position, etc.); this constant influx of information is subject to modulation to enhance or diminish stimuli (see also: gate control theory and gain control-biological). Because there are essentially unlimited stimuli, it is imperative that these signals are appropriately filtered. To diminish certain stimuli, primary afferents receive inhibitory input (likely from GABA, but could also be glycine[2]) which reduce their likelihood of synaptic output. There is evidence to support the hypothesis that presynaptic inhibition functions as an analgesic to relieve pain. When firing of nociceptive (pain-sensing) neurons is reduced, this will also reduce pain perception. One study showed that animals without a specific type of GABA receptor on their nociceptors were hypersensitive to pain,[3] thus supporting an function as an analgesic. In addition to dampening pain, impaired presynaptic inhibition has been implicated in many neurological disorders, such as epilepsy, autism, and fragile-X syndrome.[4][5][6][7][8]
Mechanisms of Presynaptic Inhibition & Primary Afferent Depolarization (PAD)[]
Primary sensory afferents contain GABA receptors along their terminals (reviewed in:,[9] Table 1). GABA receptors are ligand-gated chloride channels, formed by the assembly of five GABA receptor subunits. In addition to the presence of GABA receptors along sensory afferent axons, the presynaptic terminal also has a distinct ionic composition that is high in chloride concentration. This is due to cation-chloride cotransporters (for example, NKCC1) that maintain highs intracellular chloride.[10]
Typically when GABA receptors are activated, it causes a chloride influx, which hyperpolarizes the cell. However, in primary afferent fibers, due to the high concentration of chloride at the presynaptic terminal and thus its altered reversal potential, GABA receptor activation actually results in a chloride efflux, and thus a resulting depolarization. This phenomenon is called primary afferent depolarization (PAD).[11][12] The GABA-induced depolarized potential at afferent axons has been demonstrated in many animals from cats to insects. Interestingly, despite the depolarized potential, GABA receptor activation along the axon still results in a reduction of neurotransmitter release and thus still is inhibitory.
There are four hypotheses which propose mechanisms behind this paradox:
- The depolarized membrane causes inactivation of voltage-gated sodium channels on the terminals and therefore the action potential is prevented from propagating.[9][13][14]
- Open GABA receptor channels act as a shunt, whereby current is dissipated of instead of being propagated to the terminals.[9][13][14][15][16][17][18][19][20]
- The depolarized membrane causes inactivation of voltage-gated calcium channels, preventing calcium influx at the synapse (which is imperative for neurotransmission).[9][14][16][17][21]
- The depolarization at the terminals generates an antidromic spike (i.e. an action potential generated in the axon and travels towards the soma), which would prevent orthodromic spikes (i.e. an action potential traveling from the cell's soma toward the axon terminals) from propagating.[15]
History of the discovery of presynaptic inhibition[]
1933: Grasser & Graham observed depolarization that originated in the sensory axon terminals[22]
1938: Baron & Matthews observed depolarization that originated in sensory axon terminals and the ventral root[23]
1957: Frank & Fuortes coined the term "presynaptic inhibition" [24]
1961: Eccles, Eccles, & Magni determined that the Dorsal Root Potential (DRP) originated from depolarization in sensory axon terminals [25]
References[]
- ^ McGann JP (2013). "Presynaptic Inhibition of Olfactory Sensory Neurons: New Mechanisms and Potential Functions". Chem Senses. 38 (6): 459–74. doi:10.1093/chemse/bjt018. PMC 3685425. PMID 23761680.
- ^ Geiman EJ, Zheng W, Fritschy JM, Alvarez FJ (2002). "Glycine and GABAA receptor subunits on Renshaw cells: Relationship with presynaptic neurotransmitters and postsynaptic gephyrin clusters". The Journal of Comparative Neurology. 444 (3): 275–289. doi:10.1002/cne.10148. PMID 11840480. S2CID 1750346.
- ^ Price, Theodore J; Cervero, Fernando; Gold, Michael S; Hammond, Donna L; Prescott, Steven A (April 2009). "Chloride Regulation in the Pain Pathway". Brain Research Reviews. 60 (1): 149–170. doi:10.1016/j.brainresrev.2008.12.015. ISSN 0165-0173. PMC 2903433. PMID 19167425.
- ^ Deidda G, Bozarth IF, Cancedda L (2014). "Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives". Frontiers in Cellular Neuroscience. 8: 119. doi:10.3389/fncel.2014.00119. PMC 4033255. PMID 24904277.
- ^ Zeilhofer HU, Wildner H, Yévenes GE (2012). "Fast synaptic inhibition in spinal sensory processing and pain control". Physiological Reviews. 92 (1): 193–235. doi:10.1152/physrev.00043.2010. PMC 3590010. PMID 22298656.
- ^ Lee E, Lee J, Kim E (2017). "Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders". Biol. Psychiatry. 81 (10): 838–847. doi:10.1016/j.biopsych.2016.05.011. PMID 27450033.
- ^ D’Hulst C, Kooy HF (2007). "The GABAA receptor: a novel target for treatment of fragile X?". Trends Neurosci. 30 (8): 425–31. doi:10.1016/j.tins.2007.06.003. PMID 17590448. S2CID 7340813.
- ^ Benarroch EE (2007). "GABAA receptor heterogeneity, function, and implications for epilepsy". Neurology. 68 (8): 612–4. doi:10.1212/01.wnl.0000255669.83468.dd. PMID 17310035. S2CID 11101571.
- ^ Jump up to: a b c d Guo D, Hu J (2014). "Spinal Presynaptic Inhibition in Pain Control". Neuroscience. 283: 95–106. doi:10.1016/j.neuroscience.2014.09.032. PMID 25255936.
- ^ Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB (2008). "Roles of cation-chloride cotransporters in neurological disease". Nat Clin Pract Neurol. 4 (9): 490–503. doi:10.1038/ncpneuro0883. PMID 18769373. S2CID 15424963.
- ^ Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA (2009). "Chloride regulation in the pain pathway". Brain Res Rev. 60 (1): 149–170. doi:10.1016/j.brainresrev.2008.12.015. PMC 2903433. PMID 19167425.
- ^ Willis WD (1999). "Dorsal root potentials and dorsal root reflexes: a double-edged sword". Exp Brain Res. 124 (4): 395–421. doi:10.1007/s002210050637. PMID 10090653. S2CID 40738560.
- ^ Jump up to: a b Cattaert, D.; El Manira, A. (1999-07-15). "Shunting versus inactivation: analysis of presynaptic inhibitory mechanisms in primary afferents of the crayfish". The Journal of Neuroscience. 19 (14): 6079–6089. doi:10.1523/JNEUROSCI.19-14-06079.1999. ISSN 1529-2401. PMC 6783106. PMID 10407044.
- ^ Jump up to: a b c Willis, William D. (2006-02-01). "John Eccles' studies of spinal cord presynaptic inhibition". Progress in Neurobiology. 78 (3–5): 189–214. doi:10.1016/j.pneurobio.2006.02.007. ISSN 0301-0082. PMID 16650518. S2CID 38669996.
- ^ Jump up to: a b Cattaert, D.; Libersat, F.; El Manira A, A. (2001-02-01). "Presynaptic inhibition and antidromic spikes in primary afferents of the crayfish: a computational and experimental analysis". The Journal of Neuroscience. 21 (3): 1007–1021. doi:10.1523/JNEUROSCI.21-03-01007.2001. ISSN 1529-2401. PMC 6762302. PMID 11157086.
- ^ Jump up to: a b Panek, Izabela; French, Andrew S.; Seyfarth, Ernst-August; Sekizawa, Shin-ichi; Torkkeli, Päivi H. (July 2002). "Peripheral GABAergic inhibition of spider mechanosensory afferents". The European Journal of Neuroscience. 16 (1): 96–104. doi:10.1046/j.1460-9568.2002.02065.x. ISSN 0953-816X. PMID 12153534. S2CID 20750558.
- ^ Jump up to: a b French, Andrew S.; Panek, Izabela; Torkkeli, Päivi H. (June 2006). "Shunting versus inactivation: simulation of GABAergic inhibition in spider mechanoreceptors suggests that either is sufficient". Neuroscience Research. 55 (2): 189–196. doi:10.1016/j.neures.2006.03.002. ISSN 0168-0102. PMID 16616790. S2CID 2099107.
- ^ Miller, R. J. (1998). "Presynaptic receptors". Annual Review of Pharmacology and Toxicology. 38: 201–227. doi:10.1146/annurev.pharmtox.38.1.201. ISSN 0362-1642. PMID 9597154.
- ^ Zhang, S. J.; Jackson, M. B. (March 1995). "Properties of the GABAA receptor of rat posterior pituitary nerve terminals". Journal of Neurophysiology. 73 (3): 1135–1144. doi:10.1152/jn.1995.73.3.1135. ISSN 0022-3077. PMID 7608760.
- ^ Zhang, S. J.; Jackson, M. B. (1995-03-15). "GABAA receptor activation and the excitability of nerve terminals in the rat posterior pituitary". The Journal of Physiology. 483 ( Pt 3) (3): 583–595. doi:10.1113/jphysiol.1995.sp020608. ISSN 0022-3751. PMC 1157804. PMID 7776245.
- ^ Graham, B.; Redman, S. (February 1994). "A simulation of action potentials in synaptic boutons during presynaptic inhibition". Journal of Neurophysiology. 71 (2): 538–549. doi:10.1152/jn.1994.71.2.538. ISSN 0022-3077. PMID 8176423.
- ^ Gasser & Graham (1933). "Potentials produced in the spinal cord by stimulation of dorsal roots". American Journal of Physiology. 103 (2): 303–320. doi:10.1152/ajplegacy.1933.103.2.303.
- ^ Barron & Matthews (1938). "The Interpretation of Potential Changes in the Spinal Cord". Journal of Physiology. 92 (3): 276–321. doi:10.1113/jphysiol.1938.sp003603. PMC 1395290. PMID 16994975.
- ^ Frank & Fuortes (1957). "Presynaptic and Postsynaptic inhibition of monsynaptic reflexes". Federation Proceedings. 16: 39–40.
- ^ Eccles, Eccles, & Magni (1961). "Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys". Journal of Physiology (London). 159: 147–166. doi:10.1113/jphysiol.1961.sp006798. PMC 1359583. PMID 13889050.CS1 maint: multiple names: authors list (link)
- Neurology