Single-cell analysis
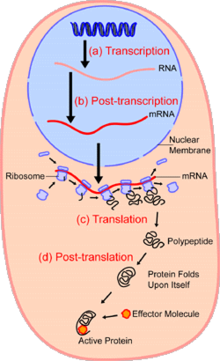
In the field of cellular biology, single-cell analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level.[1][2][3] Due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells.[4] Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies,[5][6][7] enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells,[8] as well as to quantify their proteome and metabolome.[9][10][11] Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells.[12][13] Recent advances have enabled quantifying thousands of protein across hundreds of single cells,[14] and thus make possible new types of analysis.[15][16] In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.[17]
Single-cell isolation[]
Many single-cell analysis techniques require the isolation of individual cells. Methods currently used for single cell isolation include: Dielectrophoretic digital sorting, enzymatic digestion, FACS, hydrodynamic traps, laser capture microdissection, manual picking, microfluidics, micromanipulation, serial dilution, and Raman tweezers.
Manual single cell picking is a method where cells in a suspension are viewed under a microscope, and individually picked using a micropipette.[18][19] Raman tweezers is a technique where Raman spectroscopy is combined with optical tweezers, which uses a laser beam to trap, and manipulate cells.[20]
The Dielectrophoretic digital sorting method utilizes a semiconductor controlled array of electrodes in a microfluidic chip to trap single cells in Dielectrophoretic (DEP) cages. Cell identification is ensured by the combination of fluorescent markers with image observation. Precision delivery is ensured by the semiconductor controlled motion of DEP cages in the flow cell.
The development of hydrodynamic-based microfluidic biochips has been increasing over the years. In this technique, the cells or particles are trapped in a particular region for single cell analysis (SCA) usually without any application of external force fields such as optical, electrical, magnetic or acoustic. There is a need to explore the insights of SCA in the cell's natural state and development of these techniques is highly essential for that study. Researchers have highlighted the vast potential field that needs to be explored to develop biochip devices to suit market/researcher demands. Hydrodynamic microfluidics facilitates the development of passive lab-on-chip applications. A latest review gives an account of the recent advances in this field, along with their mechanisms, methods and applications.[21]
Associated Technologies[]
Dielectrophoretic digital sorting method utilizes a semiconductor controlled array of electrodes in a microfluidic chip to trap single cells in Dielectrophoretic (DEP) cages. Cell identification is ensured by the combination of fluorescent markers with image observation. Precision delivery is ensured by the semiconductor controlled motion of DEP cages in the flow cell.
Hydrodynamic traps allow for the isolation of an individual cell in a "trap" at a single given time by passive microfluidic transport. The number of isolated cells can be manipulated based on the number of traps in the system.
The Laser Capture Microdissection technique utilizes a laser to dissect and separate individual cells, or sections, from tissue samples of interest. The methods involve the observation of a cell under a microscope, so that a section for analysis can be identified and labeled so that the laser can cut the cell. Then, the cell can be extracted for analysis.
Manual single cell picking is a method where cells in a suspension are viewed under a microscope and individually picked using a micropipette.
Microfluidics allows for the isolation of individual cells for further analyses. The following principles outline the various microfluidic processes for single-cell separation: droplet-in-oil based isolation, pneumatic membrane valving, and hydrodynamic cell traps. Droplet-in-oil based microfluidics uses oil-filled channels to hold separated aqueous droplets. This allows the single cell to be contained and isolated from the inside the oil based channels. Pneumatic membrane valves use the manipulation of air pressure, to isolate individual cells by membrane deflection. The manipulation of the pressure source allows the opening or closing of channels in a microfluidic network. Typically, the system requires an operator and is limited in throughput.
The technique Raman tweezers combines the use Raman spectroscopy and optical tweezers, which use a laser beam to trap and manipulate cells.
The development of hydrodynamic-based microfluidic biochips has been increasing over the years. In this technique, the cells are trapped in a particular region for single cell analysis (SCA). This usually occurs without any application of external force fields such as optical, electrical, magnetic or acoustic. There is a need to explore the insights of SCA in the cell's natural state, and development of these techniques is highly essential for that study. Researchers have highlighted the need to develop biochip devices to suit market and researcher demands. Hydrodynamic microfluidics facilitate the development of passive lab-on-chip applications.
Genomics[]
Techniques[]
Single-cell genomics is heavily dependent on increasing the copies of DNA found in the cell so there is enough to be sequenced. This has led to the development of strategies for whole genome amplification (WGA). Currently WGA strategies can be grouped into three categories:
- Controlled priming and PCR Amplification: Adapter-Linker PCR WGA
- Random priming and PCR Amplification: DOP-PCR, MALBAC
- Random priming and isothermal amplification: MDA
The Adapter Linker PCR WGA is reported in many comparative studies to be best performing for diploid single cell mutation analysis, thanks to its very low Allelic Dropout effect,[22][23][24] and for copy number variation profiling due to its low noise, both with aCGH and with NGS low Pass Sequencing.[25][26] This method is only applicable to human cells, both fixed and unfixed.
One widely adopted WGA techniques is called degenerate oligonucleotide–primed polymerase chain reaction (DOP-PCR). This method uses the well established DNA amplification method PCR to try and amplify the entire genome using a large set of primers. Although simple, this method has been shown to have very low genome coverage. An improvement on DOP-PCR is Multiple displacement amplification (MDA), which uses random primers and a high fidelity enzyme, usually Φ29 DNA polymerase, to accomplish the amplification of larger fragments and greater genome coverage than DOP-PCR. Despite these improvement MDA still has a sequence dependent bias (certain parts of the genome are amplified more than others because of their sequence). The method shown to largely avoid the bias seen in DOP-PCR and MDA is Multiple Annealing and Looping–Based Amplification Cycles (MALBAC). Bias in this system is reduced by only copying off the original DNA strand instead of making copies of copies. The main draw backs to using MALBA, is it has reduced accuracy compared to DOP-PCR and MDA due to the enzyme used to copy the DNA.[9] Once amplified using any of the above techniques, the DNA can be sequenced using Sanger or next-generation sequencing (NGS).
Purpose[]
There are two major applications to studying the genome at the single cell level. One application is to track the changes that occur in bacterial populations, where phenotypic differences are often seen. These differences are missed by bulk sequencing of a population, but can be observed in single cell sequencing.[27] The second major application is to study the genetic evolution of cancer. Since cancer cells are constantly mutating it is of great interest to see how cancers evolve at the genetic level. These patterns of somatic mutations and copy number aberration can be observed using single cell sequencing.[1]
Transcriptomics[]
Techniques[]
Single-cell transcriptomics uses sequencing techniques similar to single cell genomics or direct detection using fluorescence in situ hybridization. The first step in quantifying the transcriptome is to convert RNA to cDNA using reverse transcriptase so that the contents of the cell can be sequenced using NGS methods as was done in genomics. Once converted, there is not enough cDNA to be sequenced so the same DNA amplification techniques discussed in single cell genomics are applied to the cDNA to make sequencing possible.[1] Alternately, fluorescent compounds attached to RNA hybridization probes are used to identify specific sequences and sequential application of different RNA probes will build up a comprehensive transcriptome.[28][29]
Purpose[]
The purpose of single cell transcriptomics is to determine what genes are being expressed in each cell. The transcriptome is often used to quantify the gene expression instead of the proteome because of the difficulty currently associated with amplifying protein levels.[1]
There are three major reasons gene expression has been studied using this technique: to study gene dynamics, RNA splicing, and cell typing. Gene dynamics are usually studied to determine what changes in gene expression effect different cell characteristics. For example, this type of transcriptomic analysis has often been used to study embryonic development. RNA splicing studies are focused on understanding the regulation of different transcript isoforms. Single cell transcriptomics has also been used for cell typing, where the genes expressed in a cell are used to identify types of cells. The main goal in cell typing is to find a way to determine the identity of cells that don't have known genetic markers.[1]
Proteomics[]
Techniques[]
There are three major approaches to single-cell proteomics: antibody based methods, fluorescent protein based methods, and mass-spectroscopy based methods.[30][16]
Antibody–based methods[]
The antibody based methods use designed antibodies to bind to proteins of interest, allowing the relative abundance of multiple individual targets to be identified by one of several different techniques.
Imaging: Antibodies can be bound to fluorescent molecules such as quantum dots or tagged with organic fluorophores for detection by fluorescence microscopy. Since different colored quantum dots or unique fluorophores are attached to each antibody it is possible to identify multiple different proteins in a single cell. Quantum dots can be washed off of the antibodies without damaging the sample, making it possible to do multiple rounds of protein quantification using this method on the same sample.[31] For the methods based on organic fluorophores, the fluorescent tags are attached by a reversible linkage such as a DNA-hybrid (that can be melted/dissociated under low-salt conditions)[32] or chemically inactivated,[33] allowing multiple cycles of analysis, with 3-5 targets quantified per cycle. These approaches have been used for quantifying protein abundance in patient biopsy samples (e.g. cancer) to map variable protein expression in tissues and/or tumors,[33] and to measure changes in protein expression and cell signaling in response to cancer treatment.[32]
Mass Cytometry: rare metal isotopes, not normally found in cells or tissues, can be attached to the individual antibodies and detected by mass spectrometry for simultaneous and sensitive identification of proteins.[34] These techniques can be highly multiplexed for simultaneous quantification of many targets (panels of up to 38 markers) in single cells.[35]
Antibody-DNA quantification: another antibody-based method converts protein levels to DNA levels.[30] The conversion to DNA makes it possible to amplify protein levels and use NGS to quantify proteins. In one such approach, two antibodies are selected for each protein needed to be quantified. The two antibodies are then modified to have single stranded DNA connected to them that are complementary. When the two antibodies bind to a protein the complementary strands will anneal and produce a double stranded segment of DNA that can then be amplified using PCR. Each pair of antibodies designed for one protein is tagged with a different DNA sequence. The DNA amplified from PCR can then be sequenced, and the protein levels quantified.[36]
Mass spectroscopy–based methods[]
In mass spectroscopy based proteomics there are three major steps needed for peptide identification: sample preparation, separation of peptides, and identification of peptides. Several groups have focused on oocytes or very early cleavage-stage cells since these cells are unusually large and provide enough material for analysis.[37][38][39] Another approach, single cell proteomics by mass spectrometry (SCoPE-MS) has quantified thousands of proteins in mammalian cells with typical cell sizes (diameter of 10-15 μm) by combining carrier-cells and single-cell barcoding.[40][41][42][43] The second generation SCoPE-MS,[44] SCoPE2,[45][46] increased the throughput by automated and miniaturized sample preparation;[43] one such approach is MAMS (Micro-Arrays for Mass Spectrometry), which achieves aliquoting at high speeds by exploiting differences in wettability between recipient sites and surrounding areas.[47] It also improved quantitative reliability and proteome coverage by data-driven optimization of LC-MS/MS[48] and peptide identification.[49] Multiple methods exist to isolate the peptides for analysis. These include using filter aided sample preparation, the use of magnetic beads, or using a series of reagents and centrifuging steps.[50][37][39] The separation of differently sized proteins can be accomplished by using capillary electrophoresis (CE) or liquid chromatography (LC) (using liquid chromatography with mass spectroscopy is also known as LC-MS).[37][38][39][40] This step gives order to the peptides before quantification using tandem mass-spectroscopy (MS/MS). The major difference between quantification methods is some use labels on the peptides such as tandem mass tags (TMT) or dimethyl labels which are used to identify which cell a certain protein came from (proteins coming from each cell have a different label) while others use not labels (quantify cells individually). The mass spectroscopy data is then analyzed by running data through databases that convert the information about peptides identified to quantification of protein levels.[37][38][39][40][51] These methods are very similar to those used to quantify the proteome of bulk cells, with modifications to accommodate the very small sample volume.[41]
Purpose[]
The purpose of studying the proteome is to better understand the activity of cells at the single cells level. Since proteins are responsible for determining how the cell acts, understanding the proteome of single cell gives the best understanding of how a cell operates, and how gene expression changes in a cell due to different environmental stimuli. Although transcriptomics has the same purpose as proteomics it is not as accurate at determining gene expression in cells as it does not take into account post-transcriptional regulation.[10] Transcriptomics is still important as studying the difference between RNA levels and protein levels could give insight on which genes are post-transcriptionally regulated.
Metabolomics[]
Techniques[]
There are four major methods used to quantify the metabolome of single cells, they are: fluorescence–based detection, fluorescence biosensors, FRET biosensors, and mass spectroscopy. The first three methods listed use fluorescence microscopy to detect molecules in a cell. Usually these assays use small fluorescent tags attached to molecules of interest, however this has been shown be too invasive for single cell metabolomics, and alters the activity of the metabolites. The current solution to this problem is to use fluorescent proteins which will act as metabolite detectors, fluorescing when ever they bind to a metabolite of interest.[52]
Mass spectroscopy is becoming the most frequently used method for single cell metabolomics. Its advantages are that there is no need to develop fluorescent proteins for all molecules of interest, and is capable of detecting metabolites in the femtomole range.[13] Similar to the methods discussed in proteomics, there has also been success in combining mass spectroscopy with separation techniques such as capillary electrophoresis to quantify metabolites. This method is also capable of detecting metabolites present in femtomole concentrations.[52] Another method utilizing capillary microsampling combined with mass spectrometry with ion mobility separation has been demonstrated to enhance the molecular coverage and ion separation for single cell metabolomics.[19][53] Researchers are trying to develop a technique that can fulfil what current techniques are lacking: high throughput, higher sensitivity for metabolites that have a lower abundance or that have low ionization efficiencies, good replicability and that allow quantification of metabolites.[54]
Purpose[]
The purpose of single cell metabolomics is to gain a better understanding at the molecular level of major biological topics such as: cancer, stem cells, aging, as well as the development of drug resistance. In general the focus of metabolomics is mostly on understanding how cells deal with environmental stresses at the molecular level, and to give a more dynamic understanding of cellular functions.[52]
Reconstructing developmental trajectories[]
Single-cell transcriptomic assays have allowed reconstruction development trajectories. Branching of these trajectories describes cell differentiation. Various methods have been developed for reconstructing branching developmental trajectories from single-cell transcriptomic data.[55][56][57][58] They use various advanced mathematical concepts from optimal transportation[57] to principal graphs.[58] Some software libraries for reconstruction and visualization of lineage differentiation trajectories are freely available online.[59]
Cell–cell interaction[]
Cell–cell interactions are characterized by stable and transient interactions.
See also[]
- Cell sorting
- Micro-arrays for mass spectrometry
- Single-cell variability
References[]
- ^ a b c d e Wang D, Bodovitz S (June 2010). "Single cell analysis: the new frontier in 'omics'". Trends in Biotechnology. 28 (6): 281–90. doi:10.1016/j.tibtech.2010.03.002. PMC 2876223. PMID 20434785.
- ^ Habibi I, Cheong R, Lipniacki T, Levchenko A, Emamian ES, Abdi A (April 2017). "Computation and measurement of cell decision making errors using single cell data". PLOS Computational Biology. 13 (4): e1005436. Bibcode:2017PLSCB..13E5436H. doi:10.1371/journal.pcbi.1005436. PMC 5397092. PMID 28379950.
- ^ Merouane A, Rey-Villamizar N, Lu Y, Liadi I, Romain G, Lu J, et al. (October 2015). "Automated profiling of individual cell-cell interactions from high-throughput time-lapse imaging microscopy in nanowell grids (TIMING)". Bioinformatics. 31 (19): 3189–97. doi:10.1093/bioinformatics/btv355. PMC 4693004. PMID 26059718.
- ^ Altschuler SJ, Wu LF (May 2010). "Cellular heterogeneity: do differences make a difference?". Cell. 141 (4): 559–63. doi:10.1016/j.cell.2010.04.033. PMC 2918286. PMID 20478246.
- ^ Hu P, Zhang W, Xin H, Deng G (2016-10-25). "Single Cell Isolation and Analysis". Frontiers in Cell and Developmental Biology. 4: 116. doi:10.3389/fcell.2016.00116. PMC 5078503. PMID 27826548.
- ^ Mora-Castilla S, To C, Vaezeslami S, Morey R, Srinivasan S, Dumdie JN, et al. (August 2016). "Miniaturization Technologies for Efficient Single-Cell Library Preparation for Next-Generation Sequencing". Journal of Laboratory Automation. 21 (4): 557–67. doi:10.1177/2211068216630741. PMC 4948133. PMID 26891732.
- ^ Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. (January 2017). "Massively parallel digital transcriptional profiling of single cells". Nature Communications. 8: 14049. Bibcode:2017NatCo...814049Z. doi:10.1038/ncomms14049. PMC 5241818. PMID 28091601.
- ^ Mercatelli D, Balboni N, Palma A, Aleo E, Sanna PP, Perini G, Giorgi FM (January 2021). "Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines". Biomolecules. 11 (2): 177. doi:10.3390/biom11020177. PMC 7912277. PMID 33525507.
- ^ a b Huang L, Ma F, Chapman A, Lu S, Xie XS (2015). "Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications". Annual Review of Genomics and Human Genetics. 16 (1): 79–102. doi:10.1146/annurev-genom-090413-025352. PMID 26077818. S2CID 12987987.
- ^ a b Wu AR, Wang J, Streets AM, Huang Y (June 2017). "Single-Cell Transcriptional Analysis". Annual Review of Analytical Chemistry. 10 (1): 439–462. doi:10.1146/annurev-anchem-061516-045228. PMID 28301747. S2CID 40069109.
- ^ Tsioris K, Torres AJ, Douce TB, Love JC (2014). "A new toolbox for assessing single cells". Annual Review of Chemical and Biomolecular Engineering. 5: 455–77. doi:10.1146/annurev-chembioeng-060713-035958. PMC 4309009. PMID 24910919.
- ^ Comi TJ, Do TD, Rubakhin SS, Sweedler JV (March 2017). "Categorizing Cells on the Basis of their Chemical Profiles: Progress in Single-Cell Mass Spectrometry". Journal of the American Chemical Society. 139 (11): 3920–3929. doi:10.1021/jacs.6b12822. PMC 5364434. PMID 28135079.
- ^ a b Zhang L, Vertes A (April 2018). "Single-Cell Mass Spectrometry Approaches to Explore Cellular Heterogeneity". Angewandte Chemie. 57 (17): 4466–4477. doi:10.1002/anie.201709719. PMID 29218763. S2CID 4928231.
- ^ Slavov N (June 2020). "Single-cell protein analysis by mass spectrometry". Current Opinion in Chemical Biology. 60: 1–9. arXiv:2004.02069. doi:10.1016/j.cbpa.2020.04.018. ISSN 1367-5931. PMC 7767890. PMID 32599342. S2CID 219966629.
- ^ Specht H, Slavov N (August 2018). "Transformative Opportunities for Single-Cell Proteomics". Journal of Proteome Research. 17 (8): 2565–2571. doi:10.1021/acs.jproteome.8b00257. PMC 6089608. PMID 29945450.
- ^ a b Slavov N (January 2020). "Unpicking the proteome in single cells". Science. 367 (6477): 512–513. Bibcode:2020Sci...367..512S. doi:10.1126/science.aaz6695. PMC 7029782. PMID 32001644.
- ^ Lee JH (July 2017). "De Novo Gene Expression Reconstruction in Space". Trends in Molecular Medicine. 23 (7): 583–593. doi:10.1016/j.molmed.2017.05.004. PMC 5514424. PMID 28571832.
- ^ Gross A, Schoendube J, Zimmermann S, Steeb M, Zengerle R, Koltay P (July 2015). "Technologies for Single-Cell Isolation". International Journal of Molecular Sciences. 16 (8): 16897–919. doi:10.3390/ijms160816897. PMC 4581176. PMID 26213926.
- ^ a b Zhang L, Vertes A (October 2015). "Energy Charge, Redox State, and Metabolite Turnover in Single Human Hepatocytes Revealed by Capillary Microsampling Mass Spectrometry". Analytical Chemistry. 87 (20): 10397–405. doi:10.1021/acs.analchem.5b02502. PMID 26398405.
- ^ Faria EC, Gardner P (January 2012). "Analysis of single eukaryotic cells using Raman Tweezers". In Lindström S, Andersson-Svahn H (eds.). Single-Cell Analysis. Methods in Molecular Biology. Vol. 853. Humana Press. pp. 151–67. doi:10.1007/978-1-61779-567-1_12. ISBN 978-1-61779-566-4. PMID 22323146.
- ^ Narayanamurthy V, Nagarajan S, Samsuri F, Sridhar TM (2017-06-30). "Microfluidic hydrodynamic trapping for single cell analysis: mechanisms, methods and applications". Analytical Methods. 9 (25): 3751–3772. doi:10.1039/C7AY00656J. ISSN 1759-9679.
- ^ Babayan A, Alawi M, Gormley M, Müller V, Wikman H, McMullin RP, et al. (August 2017). "Comparative study of whole genome amplification and next generation sequencing performance of single cancer cells". Oncotarget. 8 (34): 56066–56080. doi:10.18632/oncotarget.10701. PMC 5593545. PMID 28915574.
- ^ Binder V, Bartenhagen C, Okpanyi V, Gombert M, Moehlendick B, Behrens B, et al. (October 2014). "A new workflow for whole-genome sequencing of single human cells". Human Mutation. 35 (10): 1260–70. doi:10.1002/humu.22625. PMID 25066732. S2CID 27392899.
- ^ Borgström E, Paterlini M, Mold JE, Frisen J, Lundeberg J (2017). "Comparison of whole genome amplification techniques for human single cell exome sequencing". PLOS ONE. 12 (2): e0171566. Bibcode:2017PLoSO..1271566B. doi:10.1371/journal.pone.0171566. PMC 5313163. PMID 28207771.
- ^ Normand E, Qdaisat S, Bi W, Shaw C, Van den Veyver I, Beaudet A, Breman A (September 2016). "Comparison of three whole genome amplification methods for detection of genomic aberrations in single cells". Prenatal Diagnosis. 36 (9): 823–30. doi:10.1002/pd.4866. PMID 27368744. S2CID 5537482.
- ^ Vander Plaetsen AS, Deleye L, Cornelis S, Tilleman L, Van Nieuwerburgh F, Deforce D (December 2017). "STR profiling and Copy Number Variation analysis on single, preserved cells using current Whole Genome Amplification methods". Scientific Reports. 7 (1): 17189. Bibcode:2017NatSR...717189V. doi:10.1038/s41598-017-17525-5. PMC 5719346. PMID 29215049.
- ^ Kalisky T, Quake SR (April 2011). "Single-cell genomics". Nature Methods. 8 (4): 311–4. doi:10.1038/nmeth0411-311. PMID 21451520. S2CID 5601612.
- ^ Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L (April 2014). "Single-cell in situ RNA profiling by sequential hybridization". Nature Methods. 11 (4): 360–1. doi:10.1038/nmeth.2892. PMC 4085791. PMID 24681720.
- ^ Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X (April 2015). "RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells" (PDF). Science. 348 (6233): aaa6090. doi:10.1126/science.aaa6090. PMC 4662681. PMID 25858977.
- ^ a b Levy E, Slavov N (October 2018). "Single cell protein analysis for systems biology". Essays in Biochemistry. 62 (4): 595–605. doi:10.1042/EBC20180014. PMC 6204083. PMID 30072488.
- ^ Zrazhevskiy P, True LD, Gao X (October 2013). "Multicolor multicycle molecular profiling with quantum dots for single-cell analysis". Nature Protocols. 8 (10): 1852–69. doi:10.1038/nprot.2013.112. PMC 4108347. PMID 24008381.
- ^ a b Giedt RJ, Pathania D, Carlson JC, McFarland PJ, Del Castillo AF, Juric D, Weissleder R (October 2018). "Single-cell barcode analysis provides a rapid readout of cellular signaling pathways in clinical specimens". Nature Communications. 9 (1): 4550. Bibcode:2018NatCo...9.4550G. doi:10.1038/s41467-018-07002-6. PMC 6208406. PMID 30382095.
- ^ a b Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. (July 2018). Chakraborty AK, Raj A, Marr C, Horváth P (eds.). "Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes". eLife. 7: e31657. doi:10.7554/eLife.31657. PMC 6075866. PMID 29993362.
- ^ Nair N, Mei HE, Chen SY, Hale M, Nolan GP, Maecker HT, et al. (May 2015). "Mass cytometry as a platform for the discovery of cellular biomarkers to guide effective rheumatic disease therapy". Arthritis Research & Therapy. 17: 127. doi:10.1186/s13075-015-0644-z. PMC 4436107. PMID 25981462.
- ^ Spitzer MH, Nolan GP (May 2016). "Mass Cytometry: Single Cells, Many Features". Cell. 165 (4): 780–91. doi:10.1016/j.cell.2016.04.019. PMC 4860251. PMID 27153492.
- ^ Gong H, Holcomb I, Ooi A, Wang X, Majonis D, Unger MA, Ramakrishnan R (January 2016). "Simple Method To Prepare Oligonucleotide-Conjugated Antibodies and Its Application in Multiplex Protein Detection in Single Cells". Bioconjugate Chemistry. 27 (1): 217–25. doi:10.1021/acs.bioconjchem.5b00613. PMID 26689321.
- ^ a b c d Lombard-Banek C, Reddy S, Moody SA, Nemes P (August 2016). "Label-free Quantification of Proteins in Single Embryonic Cells with Neural Fate in the Cleavage-Stage Frog (Xenopus laevis) Embryo using Capillary Electrophoresis Electrospray Ionization High-Resolution Mass Spectrometry (CE-ESI-HRMS)". Molecular & Cellular Proteomics. 15 (8): 2756–68. doi:10.1074/mcp.M115.057760. PMC 4974349. PMID 27317400.
- ^ a b c Sun L, Dubiak KM, Peuchen EH, Zhang Z, Zhu G, Huber PW, Dovichi NJ (July 2016). "Single Cell Proteomics Using Frog (Xenopus laevis) Blastomeres Isolated from Early Stage Embryos, Which Form a Geometric Progression in Protein Content". Analytical Chemistry. 88 (13): 6653–7. doi:10.1021/acs.analchem.6b01921. PMC 4940028. PMID 27314579.
- ^ a b c d Virant-Klun I, Leicht S, Hughes C, Krijgsveld J (August 2016). "Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes". Molecular & Cellular Proteomics. 15 (8): 2616–27. doi:10.1074/mcp.M115.056887. PMC 4974340. PMID 27215607.
- ^ a b c Budnik B, Levy E, Slavov N (2017-03-15). "Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation". bioRxiv 10.1101/102681.
- ^ a b "SCoPE-MS -- We can finally do single cell proteomics!!!". News in Proteomics Research. 2017-03-09. Retrieved 2017-06-28.
- ^ "Single-cell proteomics – Slavov Lab Blog". Slavov Lab Blog. 2017-06-06. Retrieved 2017-06-27.
- ^ a b Specht H, Harmange G, Perlman DH, Emmott E, Niziolek Z, Budnik B, Slavov N (2018-08-25). "Automated sample preparation for high-throughput single-cell proteomics". bioRxiv: 399774. doi:10.1101/399774.
- ^ Budnik B, Levy E, Harmange G, Slavov N (October 2018). "SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation". Genome Biology. 19 (1): 161. doi:10.1186/s13059-018-1547-5. PMC 6196420. PMID 30343672.
- ^ Specht H, Emmott E, Koller T, Slavov N (2019-07-09). "High-throughput single-cell proteomics quantifies the emergence of macrophage heterogeneity". bioRxiv. doi:10.1101/665307.
- ^ Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, et al. (January 2021). "Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2". Genome Biology. 22 (1): 50. doi:10.1186/s13059-021-02267-5. PMC 7839219. PMID 33504367.
- ^ Urban PL, Jefimovs K, Amantonico A, Fagerer SR, Schmid T, Mädler S, et al. (December 2010). "High-density micro-arrays for mass spectrometry". Lab on a Chip. 10 (23): 3206–9. doi:10.1039/C0LC00211A. PMID 20938499. S2CID 8747868.
- ^ Huffman RG, Chen A, Specht H, Slavov N (June 2019). "DO-MS: Data-Driven Optimization of Mass Spectrometry Methods". Journal of Proteome Research. 18 (6): 2493–2500. doi:10.1021/acs.jproteome.9b00039. PMC 6737531. PMID 31081635.
- ^ Chen AT, Franks A, Slavov N (July 2019). Cox J (ed.). "DART-ID increases single-cell proteome coverage". PLOS Computational Biology. 15 (7): e1007082. Bibcode:2019PLSCB..15E7082C. doi:10.1371/journal.pcbi.1007082. PMC 6625733. PMID 31260443.
- ^ Wiśniewski JR, Zougman A, Nagaraj N, Mann M (May 2009). "Universal sample preparation method for proteome analysis". Nature Methods. 6 (5): 359–62. doi:10.1038/nmeth.1322. PMID 19377485. S2CID 205418951.
- ^ Smits AH, Lindeboom RG, Perino M, van Heeringen SJ, Veenstra GJ, Vermeulen M (September 2014). "Global absolute quantification reveals tight regulation of protein expression in single Xenopus eggs". Nucleic Acids Research. 42 (15): 9880–91. doi:10.1093/nar/gku661. PMC 4150773. PMID 25056316.
- ^ a b c Zenobi R (December 2013). "Single-cell metabolomics: analytical and biological perspectives". Science. 342 (6163): 1243259. doi:10.1126/science.1243259. PMID 24311695. S2CID 21381091.
- ^ Zhang L, Foreman DP, Grant PA, Shrestha B, Moody SA, Villiers F, et al. (October 2014). "In situ metabolic analysis of single plant cells by capillary microsampling and electrospray ionization mass spectrometry with ion mobility separation". The Analyst. 139 (20): 5079–85. Bibcode:2014Ana...139.5079Z. doi:10.1039/C4AN01018C. PMID 25109271.
- ^ Duncan KD, Fyrestam J, Lanekoff I (January 2019). "Advances in mass spectrometry based single-cell metabolomics". The Analyst. 144 (3): 782–793. Bibcode:2019Ana...144..782D. doi:10.1039/C8AN01581C. PMID 30426983.
- ^ Haghverdi L, Büttner M, Wolf FA, Buettner F, Theis FJ (October 2016). "Diffusion pseudotime robustly reconstructs lineage branching" (PDF). Nature Methods. 13 (10): 845–8. doi:10.1038/nmeth.3971. PMID 27571553. S2CID 3594049.
- ^ Setty M, et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 34, 637–645 (2016).
- ^ a b Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, et al. (February 2019). "Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming". Cell. 176 (4): 928–943.e22. doi:10.1016/j.cell.2019.01.006. PMC 6402800. PMID 30712874.
- ^ a b Chen H, Albergante L, Hsu JY, Lareau CA, Lo Bosco G, Guan J, et al. (April 2019). "Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM". Nature Communications. 10 (1): 1903. Bibcode:2019NatCo..10.1903C. doi:10.1038/s41467-019-09670-4. PMC 6478907. PMID 31015418.
- ^ Pinello Lab. Single-Cell Trajectory Reconstruction Exploration and Mapping
- Analytical chemistry
- Biochemistry
- Cell biology
- Laboratory techniques
- Scientific techniques