Wallerian degeneration
| Nerve injury | |
|---|---|
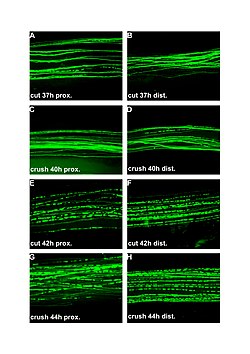 | |
| Fluorescent micrographs (100x) of Wallerian degeneration in cut and crushed peripheral nerves. Left column is proximal to the injury, right is distal. A and B: 37 hours post cut. C and D: 40 hours post crush. E and F: 42 hours post cut. G and H: 44 hours post crush. | |
| Specialty | Neurology |
Wallerian degeneration is an active process of degeneration that results when a nerve fiber is cut or crushed and the part of the axon distal to the injury (i.e. farther from the neuron's cell body) degenerates.[1] A related process of dying back or retrograde degeneration known as 'Wallerian-like degeneration' occurs in many neurodegenerative diseases, especially those where axonal transport is impaired such as ALS and Alzheimer's disease.[2] Primary culture studies suggest that a failure to deliver sufficient quantities of the essential axonal protein NMNAT2 is a key initiating event.[3][4]
Wallerian degeneration occurs after axonal injury in both the peripheral nervous system (PNS) and central nervous system (CNS). It occurs in the section of the axon distal to the site of injury and usually begins within 24–36 hours of a lesion. Prior to degeneration, the distal section of the axon tends to remain electrically excitable. After injury, the axonal skeleton disintegrates, and the axonal membrane breaks apart. Axonal degeneration is followed by degradation of the myelin sheath and infiltration by macrophages. The macrophages, accompanied by Schwann cells, serve to clear the debris from the degeneration.[5][6]
Schwann cells respond to loss of axons by extrusion of their myelin sheaths, downregulation of myelin genes, dedifferentiation and proliferation. They finally align in tubes (Büngner bands) and express surface molecules that guide regenerating fibers.[7] Within 4 days of the injury, the distal end of the portion of the nerve fiber proximal to the lesion sends out sprouts towards those tubes and these sprouts are attracted by growth factors produced by Schwann cells in the tubes. If a sprout reaches the tube, it grows into it and advances about 1 mm per day, eventually reaching and reinnervating the target tissue. If the sprouts cannot reach the tube, for instance because the gap is too wide or scar tissue has formed, surgery can help to guide the sprouts into the tubes. Regeneration is efficient in the PNS, with near complete recovery in case of lesions that occur close to the distal nerve terminal. However recovery is hardly observed at all in the spinal cord. One crucial difference is that in the CNS, including the spinal cord, myelin sheaths are produced by oligodendrocytes and not by Schwann cells.
History[]
Wallerian degeneration is named after Augustus Volney Waller. Waller experimented on frogs in 1850, by severing their glossopharyngeal and hypoglossal nerves. He then observed the distal nerves from the site of injury, which were separated from their cell bodies in the brain stem.[5] Waller described the disintegration of myelin, which he referred to as "medulla", into separate particles of various sizes. The degenerating axons formed droplets that could be stained, thus allowing for studies of the course of individual nerve fibres.
Axonal degeneration[]
Although most injury responses include a calcium influx signaling to promote resealing of severed parts, axonal injuries initially lead to acute axonal degeneration (AAD), which is rapid separation of the proximal (the part nearer the cell body) and distal ends within 30 minutes of injury.[8] After separation, dystrophic bulb structures form at both terminals and the transected membranes are sealed.[9] A brief latency phase occurs in the distal segment during which it remains electrically excitable and structurally intact.[10] Degeneration follows with swelling of the axolemma, and eventually the formation of bead-like axonal spheroids. The process takes roughly 24 hours in the PNS, and longer in the CNS. The signaling pathways leading to axolemma degeneration are currently poorly understood. However, research has shown that this AAD process is calcium–independent.[11]
Granular disintegration of the axonal cytoskeleton and inner organelles occurs after axolemma degradation. Early changes include accumulation of mitochondria in the paranodal regions at the site of injury. Endoplasmic reticulum degrades and mitochondria swell up and eventually disintegrate. The depolymerization of microtubules occurs and is soon followed by degradation of the neurofilaments and other cytoskeleton components. The disintegration is dependent on Ubiquitin and Calpain proteases (caused by influx of calcium ion), suggesting that axonal degeneration is an active process and not a passive one as previously misunderstood.[12] Thus the axon undergoes complete fragmentation. The rate of degradation is dependent on the type of injury and is also slower in the CNS than in the PNS. Another factor that affects degradation rate is the diameter of the axon: larger axons require a longer time for the cytoskeleton to degrade and thus take a longer time to degenerate.
Myelin clearance[]
Myelin is a phospholipid membrane that wraps around axons to provide them with insulation. It is produced by Schwann cells in the PNS, and by oligodendrocytes in the CNS. Myelin clearance is the next step in Wallerian degeneration following axonal degeneration. The cleaning up of myelin debris is different for PNS and CNS. PNS is much faster and efficient at clearing myelin debris in comparison to CNS, and Schwann cells are the primary cause of this difference. Another key aspect is the change in permeability of the blood-tissue barrier in the two systems. In PNS, the permeability increases throughout the distal stump, but the barrier disruption in CNS is limited to just the site of injury.[11]
Clearance in PNS[]
The response of Schwann cells to axonal injury is rapid. The time period of response is estimated to be prior to the onset of axonal degeneration. Neuregulins are believed to be responsible for the rapid activation. They activate ErbB2 receptors in the Schwann cell microvilli, which results in the activation of the mitogen-activated protein kinase (MAPK).[13] Although MAPK activity is observed, the injury sensing mechanism of Schwann cells is yet to be fully understood. The 'sensing' is followed by decreased synthesis of myelin lipids and eventually stops within 48 hrs. The myelin sheaths separate from the axons at the Schmidt-Lanterman incisures first and then rapidly deteriorate and shorten to form bead-like structures. Schwann cells continue to clear up the myelin debris by degrading their own myelin, phagocytose extracellular myelin and attract macrophages to myelin debris for further phagocytosis.[11] However, the macrophages are not attracted to the region for the first few days; hence the Schwann cells take the major role in myelin cleaning until then.
Schwann cells have been observed to recruit macrophages by release of cytokines and chemokines after sensing of axonal injury. The recruitment of macrophages helps improve the clearing rate of myelin debris. The resident macrophages present in the nerves release further chemokines and cytokines to attract further macrophages. The degenerating nerve also produce macrophage chemotactic molecules. Another source of macrophage recruitment factors is serum. Delayed macrophage recruitment was observed in B-cell deficient mice lacking serum antibodies.[11] These signaling molecules together cause an influx of macrophages, which peaks during the third week after injury. While Schwann cells mediate the initial stage of myelin debris clean up, macrophages come in to finish the job. Macrophages are facilitated by opsonins, which label debris for removal. The 3 major groups found in serum include complement, pentraxins, and antibodies. However, only complement has shown to help in myelin debris phagocytosis.[14]
Murinson et al. (2005)[15] observed that non-myelinated or myelinated Schwann cells in contact with an injured axon enter cell cycle thus leading to proliferation. Observed time duration for Schwann cell divisions were approximately 3 days after injury.[16] Possible sources of proliferation signal are attributed to the ErbB2 receptors and the ErbB3 receptors. This proliferation could further enhance the myelin cleaning rates and plays an essential role in regeneration of axons observed in PNS. Schwann cells emit growth factors that attract new axonal sprouts growing from the proximal stump after complete degeneration of the injured distal stump. This leads to possible reinnervation of the target cell or organ. However, the reinnervation is not necessarily perfect, as possible misleading occurs during reinnervation of the proximal axons to target cells.
Clearance in CNS[]
In comparison to Schwann cells, oligodendrocytes require axon signals to survive. In their developmental stages, oligodendrocytes that fail to make contact to axon and receive axon signals undergo apoptosis.[17]
Experiments in Wallerian degeneration have shown that upon injury oligodendrocytes either undergo programmed cell death or enter a state of rest. Therefore, unlike Schwann cells, oligodendrocytes fail to clean up the myelin sheaths and their debris. In experiments conducted on rats,[18] myelin sheaths were found for up to 22 months. Therefore, CNS rates of myelin sheath clearance are very slow and could possibly be the cause for hindrance in the regeneration capabilities of the CNS axons as no growth factors are available to attract the proximal axons. Another feature that results eventually is Glial scar formation. This further hinders chances for regeneration and reinnervation.
Oligodendrocytes fail to recruit macrophages for debris removal. Macrophage entry in general into CNS site of injury is very slow. In contrast to PNS, Microglia play a vital role in CNS wallerian degeneration. However, their recruitment is slower in comparison to macrophage recruitment in PNS by approximately 3 days. Further, microglia might be activated but hypertrophy, and fail to transform into fully phagocytic cells. Those microglia that do transform, clear out the debris effectively. Differentiating phagocytic microglia can be accomplished by testing for expression of Major histocompatibility complex (MHC) class I and II during wallerian degeneration.[19] The rate of clearance is very slow among microglia in comparison to macrophages. Possible source for variations in clearance rates could include lack of opsonin activity around microglia, and the lack of increased permeability in the blood–brain barrier. The decreased permeability could further hinder macrophage infiltration to the site of injury.[11]
These findings have suggested that the delay in Wallerian degeneration in CNS in comparison to PNS is caused not due to a delay in axonal degeneration, but rather is due to the difference in clearance rates of myelin in CNS and PNS.[20]
Regeneration[]
Regeneration follows degeneration. Regeneration is rapid in PNS, allowing for rates of up to 1 millimeter a day of regrowth.[21] Grafts may also be needed to allow for appropriate reinnervation. It is supported by Schwann cells through growth factors release. CNS regeneration is much slower, and is almost absent in most vertebrate species. The primary cause for this could be the delay in clearing up myelin debris. Myelin debris, present in CNS or PNS, contains several inhibitory factors. The prolonged presence of myelin debris in CNS could possibly hinder the regeneration.[22] An experiment conducted on newts, animals that have fast CNS axon regeneration capabilities, found that Wallerian degeneration of an optic nerve injury took up to 10 to 14 days on average, further suggesting that slow clearance inhibits regeneration.[23]
Schwann cells and endoneural fibroblasts in PNS[]
In healthy nerves, nerve growth factor (NGF) is produced in very small amounts. However, upon injury, NGF mRNA expression increases by five to seven-fold within a period of 14 days. Nerve fibroblasts and Schwann cells play an important role in increased expression of NGF mRNA.[24] Macrophages also stimulate Schwann cells and fibroblasts to produce NGF via macrophage-derived interleukin-1.[25] Other neurotrophic molecules produced by Schwann cells and fibroblasts together include brain-derived neurotrophic factor, glial cell line-derived neurotrophic factor, ciliary neurotrophic factor, leukemia inhibitory factor, insulin-like growth factor, and fibroblast growth factor. These factors together create a favorable environment for axonal growth and regeneration.[11] Apart from growth factors, Schwann cells also provide structural guidance to further enhance regeneration. During their proliferation phase, Schwann cells begin to form a line of cells called Bands of Bungner within the basal laminar tube. Axons have been observed to regenerate in close association to these cells.[26] Schwann cells upregulate the production of cell surface adhesion molecule ninjurin further promoting growth.[27] These lines of cell guide the axon regeneration in proper direction. The possible source of error that could result from this is possible mismatching of the target cells as discussed earlier.
Due to lack of such favorable promoting factors in CNS, regeneration is stunted in CNS.
Wallerian degeneration slow[]
Mice belonging to the strain C57BL/Wlds have delayed Wallerian degeneration,[28] and, thus, allow for the study of the roles of various cell types and the underlying cellular and molecular processes. Current understanding of the process has been possible via experimentation on the Wlds strain of mice. The mutation occurred first in mice in Harlan-Olac, a laboratory producing animals the United Kingdom. The Wlds mutation is an autosomal-dominant mutation occurring in the mouse chromosome 4.[29][30] The gene mutation is an 85-kb tandem triplication, occurring naturally. The mutated region contains two associated genes: nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) and ubiquitination factor e4b (UBE4B). A linker region encoding 18 amino acids is also part of the mutation.[6] The protective effect of the WldS protein has been shown to be due to the NMNAT1 region's NAD+ synthesizing active site.[31]
Although the protein created localizes within the nucleus and is barely detectable in axons, studies suggest that its protective effect is due to its presence in axonal and terminal compartments.[32][33] The protection provided by the WldS protein is intrinsic to the neurons and not surrounding support cells, and is only locally protective of the axon, indicating an intracellular pathway is responsible for mediating Wallerian degeneration.[34][35]
Effects of the WldS mutation[]
The mutation causes no harm to the mouse. The only known effect is that the Wallerian degeneration is delayed by up to three weeks on average after injury of a nerve. At first, it was suspected that the Wlds mutation slows down the macrophage infiltration, but recent studies suggest that the mutation protects axons rather than slowing down the macrophages.[6] The process by which the axonal protection is achieved is poorly understood. However, studies suggest that the Wlds mutation leads to increased NMNAT1 activity, which leads to increased NAD+ synthesis.[31] This in turn activates SIRT1-dependent process within the nucleus, causing changes in gene transcription.[31] NAD+ by itself may provide added axonal protection by increasing the axon's energy resources.[36] More recent work, however, raises doubt that either NMNAT1 or NAD+ can substitute for the full length Wlds gene.[37] These authors demonstrated by both in vitro and in vivo methods that the protective effect of overexpression of NMNAT1 or the addition of NAD+ did not protect axons from degeneration. However, later studies showed that NMNAT1 is protective when combined with an axonal targeting peptide, suggesting that the key to the protection provided by WldS was the combination of NMNAT1's activity and the axonal localization provided by the N-terminal domain of the chimeric protein.[38]
The provided axonal protection delays the onset of Wallerian degeneration. Schwann cell activation should therefore be delayed, as they would not detect axonal degradation signals from ErbB2 receptors. In experiments on Wlds mutated mice, macrophage infiltration was considerably delayed by up to six to eight days.[39] However, once the axonal degradation has begun, degeneration takes its normal course, and, respective of the nervous system, degradation follows at the above-described rates. Possible effects of this late onset are weaker regenerative abilities in the mice. Studies indicate that regeneration may be impaired in WldS mice, but this is likely a result of the environment being unfavorable for regeneration due to the continued existence of the undegenerated distal fiber, whereas normally debris is cleared, making way for new growth.[40]
SARM1[]
The Wallerian degeneration pathway has been further illuminated by the discovery that sterile alpha and TIR motif containing 1 (SARM1) protein plays a central role in the Wallerian degeneration pathway. The gene was first identified in a Drosophila melanogaster mutagenesis screen, and subsequently knockouts of its homologue in mice showed robust protection of transected axons comparable to that of WldS.[41][42]
SARM1 catalyzes the synthesis and hydrolysis of cyclic ADP-ribose (cADPR) from NAD+ to ADP-ribose.[43] SARM1 activation locally triggers a rapid collapse of NAD+ levels in the distal section of the injured axon, which then undergoes degeneration.[44] This collapse in NAD+ levels was later shown to be due to SARM1's TIR domain having intrinsic NAD+ cleavage activity.[45] The SARM1 protein has four domains, a mitochondrial localization signal, an auto-inhibitory N-terminus region consisting of armadillo/HEAT motifs, two sterile alpha motifs responsible for multimerization, and a C-terminus Toll/Interleukin-1 receptor that possesses enzymatic activity.[45] Activation of SARM1 is sufficient to collapse NAD+ levels and initiate the Wallerian degeneration pathway.[44]
The activity of SARM1 helps to explain the protective nature of the survival factor NMNAT2, as NMNAT enzymes have been shown to prevent SARM1-mediated depletion of NAD+.[46] This relationship is further supported by the fact that mice lacking NMNAT2, which are normally not viable, are completely rescued by SARM1 deletion, placing NMNAT2 activity upstream of SARM1.[47] Other pro-degeneration signaling pathways, such as the MAP kinase pathway, have been linked to SARM1 activation. MAPK signaling has been shown to promote the loss of NMNAT2, thereby promoting SARM1 activation, although SARM1 activation also triggers the MAP kinase cascade, indicating some form of feedback loop exists.[48][49] One explanation for the protective effect of the WldS mutation is that the NMNAT1 region, which is normally localized to the soma, substitutes for the labile survival factor NMNAT2 to prevent SARM1 activation when the N-terminal Ube4 region of the WldS protein localizes it to the axon. The fact that the enhanced survival of WldS axons is due to the slower turnover of WldS compared to NMNAT2 also helps explain why SARM1 knockout confers longer protection, as SARM1 will be completely inactive regardless of inhibitor activity whereas WldS will eventually be degraded. Possibles implications of the SARM1 pathway in regard to human health may be found in animal models which exhibit traumatic brain injury, as mice which contain Sarm1 deletions in addition to WldS show decreased axonal damage following injury.[50] Specific mutations in NMNAT2 have linked the Wallerian degeneration mechanism to two neurological diseases.
See also[]
- Axonotmesis
- Connective tissue in the peripheral nervous system
- Diffuse axonal injury
- Digestion chambers
- Nerve injury
- Neuroregeneration
- Peripheral nerve injury
- Primary and secondary brain injury
- Seddon's classification
- Spinal cord injury research
References[]
- ^ Trauma and Wallerian Degeneration, University of California, San Francisco
- ^ Coleman MP, Freeman MR (1 June 2010). "Wallerian degeneration, wld(s), and nmnat". Annual Review of Neuroscience. 33 (1): 245–67. doi:10.1146/annurev-neuro-060909-153248. PMC 5223592. PMID 20345246.
- ^ Gilley J, Coleman MP (January 2010). "Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons". PLOS Biology. 8 (1): e1000300. doi:10.1371/journal.pbio.1000300. PMC 2811159. PMID 20126265.
- ^ Brazill JM, Li C, Zhu Y, Zhai RG (2017). "NMNAT: It's an NAD + Synthase… It's a Chaperone… It's a Neuroprotector". Current Opinion in Genetics & Development. 44: 156–162. doi:10.1016/j.gde.2017.03.014. PMC 5515290. PMID 28445802.
- ^ a b Waller A (1 January 1850). "Experiments on the Section of the Glossopharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres". Philosophical Transactions of the Royal Society of London. 140: 423–429. doi:10.1098/rstl.1850.0021. JSTOR 108444.
- ^ a b c Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH (August 1998). "An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse". Proceedings of the National Academy of Sciences of the United States of America. 95 (17): 9985–90. Bibcode:1998PNAS...95.9985C. doi:10.1073/pnas.95.17.9985. PMC 21448. PMID 9707587.
- ^ Stoll G, Müller HW (April 1999). "Nerve injury, axonal degeneration and neural regeneration: basic insights". Brain Pathology. 9 (2): 313–25. doi:10.1111/j.1750-3639.1999.tb00229.x. PMC 8098499. PMID 10219748. S2CID 24140507.
- ^ Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (May 2005). "In vivo imaging of axonal degeneration and regeneration in the injured spinal cord". Nature Medicine. 11 (5): 572–7. doi:10.1038/nm1229. PMID 15821747. S2CID 25287010.
- ^ Eddleman CS, Ballinger ML, Smyers ME, Fishman HM, Bittner GD (June 1998). "Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury". The Journal of Neuroscience. 18 (11): 4029–41. doi:10.1523/JNEUROSCI.18-11-04029.1998. PMC 6792792. PMID 9592084.
- ^ Wang JT, Medress ZA, Barres BA (January 2012). "Axon degeneration: molecular mechanisms of a self-destruction pathway". The Journal of Cell Biology. 196 (1): 7–18. doi:10.1083/jcb.201108111. PMC 3255986. PMID 22232700.
- ^ a b c d e f Vargas ME, Barres BA (1 July 2007). "Why is Wallerian degeneration in the CNS so slow?". Annual Review of Neuroscience. 30 (1): 153–79. doi:10.1146/annurev.neuro.30.051606.094354. PMID 17506644.
- ^ Zimmerman UP, Schlaepfer WW (March 1984). "Multiple forms of Ca-activated protease from rat brain and muscle". The Journal of Biological Chemistry. 259 (5): 3210–8. doi:10.1016/S0021-9258(17)43282-0. PMID 6321500.
- ^ Guertin AD, Zhang DP, Mak KS, Alberta JA, Kim HA (March 2005). "Microanatomy of axon/glial signaling during Wallerian degeneration". The Journal of Neuroscience. 25 (13): 3478–87. doi:10.1523/JNEUROSCI.3766-04.2005. PMC 6724908. PMID 15800203.
- ^ Dailey AT, Vellino AM, Benthem L, Silver J, Kliot M (September 1998). "Complement depletion reduces macrophage infiltration and ctivation during Wallerian degeneration and axonal regeneration". The Journal of Neuroscience. 18 (17): 6713–22. doi:10.1523/JNEUROSCI.18-17-06713.1998. PMC 6792968. PMID 9712643.
- ^ Murinson BB, Archer DR, Li Y, Griffin JW (February 2005). "Degeneration of myelinated efferent fibers prompts mitosis in Remak Schwann cells of uninjured C-fiber afferents". The Journal of Neuroscience. 25 (5): 1179–87. doi:10.1523/JNEUROSCI.1372-04.2005. PMC 6725954. PMID 15689554.
- ^ Liu HM, Yang LH, Yang YJ (July 1995). "Schwann cell properties: 3. C-fos expression, bFGF production, phagocytosis and proliferation during Wallerian degeneration". Journal of Neuropathology and Experimental Neurology. 54 (4): 487–96. doi:10.1097/00005072-199507000-00002. PMID 7602323. S2CID 25055891.
- ^ Barres BA, Jacobson MD, Schmid R, Sendtner M, Raff MC (August 1993). "Does oligodendrocyte survival depend on axons?". Current Biology. 3 (8): 489–97. doi:10.1016/0960-9822(93)90039-Q. PMID 15335686. S2CID 39909326.
- ^ Ludwin SK (31 May 1990). "Oligodendrocyte survival in Wallerian degeneration". Acta Neuropathologica. 80 (2): 184–91. doi:10.1007/BF00308922. PMID 1697140. S2CID 36103242.
- ^ Koshinaga M, Whittemore SR (April 1995). "The temporal and spatial activation of microglia in fiber tracts undergoing anterograde and retrograde degeneration following spinal cord lesion". Journal of Neurotrauma. 12 (2): 209–22. doi:10.1089/neu.1995.12.209. PMID 7629867.
- ^ George R, Griffin JW (October 1994). "Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model". Experimental Neurology. 129 (2): 225–36. doi:10.1006/exnr.1994.1164. PMID 7957737. S2CID 40089749.
- ^ Lundy-Ekman L (2007). Neuroscience: Fundamentals for Rehabilitation (3rd ed.). Saunders. ISBN 978-1-4160-2578-8.
- ^ He Z, Koprivica V (21 July 2004). "The Nogo signaling pathway for regeneration block". Annual Review of Neuroscience. 27 (1): 341–68. doi:10.1146/annurev.neuro.27.070203.144340. PMID 15217336.
- ^ Turner JE, Glaze KA (March 1977). "The early stages of Wallerian degeneration in the severed optic nerve of the newt (Triturus viridescens)". The Anatomical Record. 187 (3): 291–310. doi:10.1002/ar.1091870303. PMID 851236. S2CID 2028827.
- ^ Heumann R, Korsching S, Bandtlow C, Thoenen H (June 1987). "Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection" (PDF). The Journal of Cell Biology. 104 (6): 1623–31. doi:10.1083/jcb.104.6.1623. PMC 2114490. PMID 3034917.
- ^ Lindholm D, Heumann R, Hengerer B, Thoenen H (November 1988). "Interleukin 1 increases stability and transcription of mRNA encoding nerve growth factor in cultured rat fibroblasts". The Journal of Biological Chemistry. 263 (31): 16348–51. doi:10.1016/S0021-9258(18)37599-9. PMID 3263368.
- ^ Thomas PK, King RH (October 1974). "The degeneration of unmyelinated axons following nerve section: an ultrastructural study". Journal of Neurocytology. 3 (4): 497–512. doi:10.1007/BF01098736. PMID 4436692. S2CID 37385200.
- ^ Araki T, Milbrandt J (August 1996). "Ninjurin, a novel adhesion molecule, is induced by nerve injury and promotes axonal growth". Neuron. 17 (2): 353–61. doi:10.1016/S0896-6273(00)80166-X. PMID 8780658. S2CID 12471778.
- ^ Perry VH, Brown MC, Tsao JW (1 October 1992). "The Effectiveness of the Gene Which Slows the Rate of Wallerian Degeneration in C57BL/Ola Mice Declines With Age". The European Journal of Neuroscience. 4 (10): 1000–2. doi:10.1111/j.1460-9568.1992.tb00126.x. PMID 12106435. S2CID 24786532.
- ^ Perry, V. H., Lunn, E. R., Brown, M. C., Cahusac, S. and Gordon, S. (1990), Evidence that the Rate of Wallerian Degeneration is Controlled by a Single Autosomal Dominant Gene. European Journal of Neuroscience, 2: 408-413. https://doi.org/10.1111/j.1460-9568.1990.tb00433.x
- ^ Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH (October 1993). "A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4". Proceedings of the National Academy of Sciences of the United States of America. 90 (20): 9717–20. Bibcode:1993PNAS...90.9717L. doi:10.1073/pnas.90.20.9717. PMC 47641. PMID 8415768.
- ^ a b c Araki T, Sasaki Y, Milbrandt J (August 2004). "Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration". Science. 305 (5686): 1010–3. Bibcode:2004Sci...305.1010A. doi:10.1126/science.1098014. PMID 15310905. S2CID 32370137.
- ^ Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP (December 2001). "Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene". Nature Neuroscience. 4 (12): 1199–206. doi:10.1038/nn770. hdl:1842/737. PMID 11770485. S2CID 8316115.
- ^ Beirowski B, Babetto E, Gilley J, Mazzola F, Conforti L, Janeckova L, Magni G, Ribchester RR, Coleman MP (January 2009). "Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo". The Journal of Neuroscience. 29 (3): 653–68. doi:10.1523/JNEUROSCI.3814-08.2009. PMC 6665162. PMID 19158292.
- ^ Glass JD, Brushart TM, George EB, Griffin JW (May 1993). "Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon". Journal of Neurocytology. 22 (5): 311–21. doi:10.1007/BF01195555. PMID 8315413. S2CID 45871975.
- ^ Adalbert R, Nógrádi A, Szabó A, Coleman MP (October 2006). "The slow Wallerian degeneration gene in vivo protects motor axons but not their cell bodies after avulsion and neonatal axotomy". The European Journal of Neuroscience. 24 (8): 2163–8. doi:10.1111/j.1460-9568.2006.05103.x. PMID 17074042. S2CID 25359698.
- ^ Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z (August 2005). "A local mechanism mediates NAD-dependent protection of axon degeneration". The Journal of Cell Biology. 170 (3): 349–55. doi:10.1083/jcb.200504028. PMC 2171458. PMID 16043516.
- ^ Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP (January 2007). "NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration". Cell Death and Differentiation. 14 (1): 116–27. doi:10.1038/sj.cdd.4401944. PMID 16645633.
- ^ Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP (October 2010). "Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo". The Journal of Neuroscience. 30 (40): 13291–304. doi:10.1523/JNEUROSCI.1189-10.2010. PMC 6634738. PMID 20926655.
- ^ Fujiki M, Zhang Z, Guth L, Steward O (July 1996). "Genetic influences on cellular reactions to spinal cord injury: activation of macrophages/microglia and astrocytes is delayed in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration". The Journal of Comparative Neurology. 371 (3): 469–84. doi:10.1002/(SICI)1096-9861(19960729)371:3<469::AID-CNE9>3.0.CO;2-0. PMID 8842900.
- ^ Brown MC, Perry VH, Hunt SP, Lapper SR (March 1994). "Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration". The European Journal of Neuroscience. 6 (3): 420–8. doi:10.1111/j.1460-9568.1994.tb00285.x. PMID 8019679. S2CID 37501852.
- ^ Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR (July 2012). "dSarm/Sarm1 is required for activation of an injury-induced axon death pathway". Science. 337 (6093): 481–4. Bibcode:2012Sci...337..481O. doi:10.1126/science.1223899. PMC 5225956. PMID 22678360.
- ^ Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J (August 2013). "Sarm1-mediated axon degeneration requires both SAM and TIR interactions". The Journal of Neuroscience. 33 (33): 13569–80. doi:10.1523/JNEUROSCI.1197-13.2013. PMC 3742939. PMID 23946415.
- ^ Lee HC, Zhao YJ (2019). "Resolving the topological enigma in Ca 2+ signaling by cyclic ADP-ribose and NAADP". Journal of Biological Chemistry. 294 (52): 19831–19843. doi:10.1074/jbc.REV119.009635. PMC 6937575. PMID 31672920.
- ^ a b Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J (April 2015). "SARM1 activation triggers axon degeneration locally via NAD⁺ destruction". Science. 348 (6233): 453–7. Bibcode:2015Sci...348..453G. doi:10.1126/science.1258366. PMC 4513950. PMID 25908823.
- ^ a b Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J (March 2017). "+ Cleavage Activity that Promotes Pathological Axonal Degeneration". Neuron. 93 (6): 1334–1343.e5. doi:10.1016/j.neuron.2017.02.022. PMC 6284238. PMID 28334607.
- ^ Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J (October 2016). "+ depletion". eLife. 5. doi:10.7554/eLife.19749. PMC 5063586. PMID 27735788.
- ^ Gilley J, Ribchester RR, Coleman MP (October 2017). "S, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy". Cell Reports. 21 (1): 10–16. doi:10.1016/j.celrep.2017.09.027. PMC 5640801. PMID 28978465.
- ^ Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M (January 2015). "Pathological axonal death through a MAPK cascade that triggers a local energy deficit". Cell. 160 (1–2): 161–76. doi:10.1016/j.cell.2014.11.053. PMC 4306654. PMID 25594179.
- ^ Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A (January 2017). "MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2". eLife. 6. doi:10.7554/eLife.22540. PMC 5241118. PMID 28095293.
- ^ Henninger N, et al. (2016). "Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1". Brain. 139 (4): 1094–1105. doi:10.1093/brain/aww001. PMC 5006226. PMID 26912636.
External links[]
- Wallerian+Degeneration at the US National Library of Medicine Medical Subject Headings (MeSH)
- Neurotrauma