6-Pyruvoyltetrahydropterin synthase deficiency
| 6-Pyruvoyltetrahydropterin synthase deficiency | |
|---|---|
| Other names | Hyperphenylalaninemia due to 6-pyruvoyltetrahydropterin synthase deficiency[1] |
 | |
| This condition is inherited in an autosomal recessive manner | |
6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency.[2] It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors (levodopa, 5-hydroxytryptophan), monoamine oxidase inhibitors, and tetrahydrobiopterin.[3] Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype/phenotype correlation and outcome of these diseases their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).[4]
Presentation[]
Movement disorders mentioned are caused by the inability to produce L-Dopa. Dopamine is a neurotransmitter which is also involved in movement. The absence of enough of this molecule causes the movement disorders in an affected individual.[citation needed]
Mechanism[]
To understand how the absence of this enzyme affects the body, we must look at the BH4 synthesis pathway. PTPS is an intermediate in this cycle and is needed to convert 7,8 - dihydroneopterin triphosphate to 6-Pyruvoyltetrahydryobiopterin. 6-Pyruvoyltetrahydryobiopterin is converted into BH4 (Tetrahydrobiopterin), but since it stops at 6-Pyruvoyltetrahydrobiopterin no BH4 is made.[5] The absence of BH4 affects the metabolism of Phenylalanine. This is the reason that PKU and PTPS deficiency share some similar symptoms. However, since BH4 is needed for much more than just the metabolism of Phenylalanine, there are other symptoms as well.[citation needed]
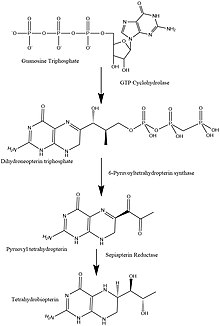
Structure and function of 6-Pyruvoyltetrahydropterin Synthase[]
PTPS is a hexamer of identical subunits. The PTPS monomer folds into a sequential four-stranded, antiparallel