H3K4me1
H3K4me1 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the mono-methylation at the 4th lysine residue of the histone H3 protein and often associated with gene enhancers.
Nomenclature[]
H3K4me1 indicates monomethylation of lysine 4 on histone H3 protein subunit: [1]
| Abbr. | Meaning |
| H3 | H3 family of histones |
| K | standard abbreviation for lysine |
| 4 | position of amino acid residue
(counting from N-terminus) |
| me | methyl group |
| 1 | number of methyl groups added |
Lysine Methylation[]
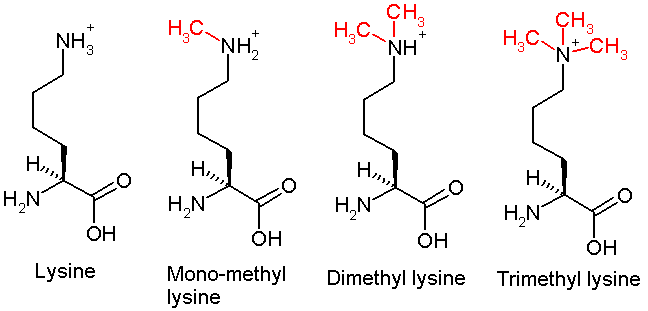
This diagram shows the progressive methylation of a lysine residue. The mono-methylation denotes the methylation present in H3K4me1.
Understanding histone modifications[]
The genomic DNA of eukaryotic cells is wrapped around special protein molecules known as Histones. The complexes formed by the looping of the DNA are known as Chromatin. The basic structural unit of chromatin is the Nucleosome: this consists of the core octamer of histones (H2A, H2B, H3 and H4) as well as a linker histone and about 180 base pairs of DNA. These core histones are rich in lysine and arginine residues. The carboxyl (C) terminal end of these histones contribute to histone-histone interactions, as well as histone-DNA interactions. The amino (N) terminal charged tails are the site of the post-translational modifications, such as the one seen in H3K4me1.[2][3]
Mechanism and function of modification[]
H3K4me1 is enriched at active and primed enhancers.[4] Transcriptional enhancers control the cell-identity gene expression and are important in the cell identity. Enhancers are primed by histone H3K4 mono-/di-methyltransferase MLL4 and then are activated by histone H3K27 acetyltransferase p300.[5] H3K4me1 fine-tunes the enhancer activity and function rather than controls.[4] H3K4me1 is put down by KMT2C (MLL3) and KMT2D (MLL4)[6]
LSD1, and the related LSD2/KDM1B demethylate H3K4me1 and H3K4me2.[7]
Marks associated with active gene transcription like H3K4me1 and H3K9me1 have very short half-lives.[8]
H3K4me1 with MLL3/4 can also act at promoters and repress genes.[8]
Relationship with other modifications[]
H3K4me1 is a chromatin signature of enhancers, H3K4me2 is highest toward the 5′ end of transcribing genes and H3K4me3 is highly enriched at promoters and in poised genes. H3K27me3, H4K20me1 and H3K4me1 silence transcription in embryonic fibroblasts, macrophages, and human embryonic stem cells (ESCs).[7]
Enhancers that have two opposing marks like the active mark H3K4me1 and repressive mark H3K27me3 at the same time are called bivalent or poised. These bivalent enhancers convert and become enriched with H3K4me1 and acetylated H3K27 (H3K27ac) after differentiation.[8]
Epigenetic implications[]
The post-translational modification of histone tails by either histone modifying complexes or chromatin remodelling complexes are interpreted by the cell and lead to complex, combinatorial transcriptional output. It is thought that a Histone code dictates the expression of genes by a complex interaction between the histones in a particular region.[9] The current understanding and interpretation of histones comes from two large scale projects: ENCODE and the Epigenomic roadmap.[10] The purpose of the epigenomic study was to investigate epigenetic changes across the entire genome. This led to chromatin states which define genomic regions by grouping the interactions of different proteins and/or histone modifications together. Chromatin states were investigated in Drosophila cells by looking at the binding location of proteins in the genome. Use of ChIP-sequencing revealed regions in the genome characterised by different banding.[11] Different developmental stages were profiled in Drosophila as well, an emphasis was placed on histone modification relevance.[12] A look in to the data obtained led to the definition of chromatin states based on histone modifications.[13] Certain modifications were mapped and enrichment was seen to localize in certain genomic regions. Five core histone modifications were found with each respective one being linked to various cell functions.
- H3K4me1- primed enhancers
- H3K4me3-promoters
- H3K36me3-gene bodies
- H3K27me3-polycomb repression
- H3K9me3-heterochromatin
The human genome was annotated with chromatin states. These annotated states can be used as new ways to annotate a genome independently of the underlying genome sequence. This independence from the DNA sequence enforces the epigenetic nature of histone modifications. Chromatin states are also useful in identifying regulatory elements that have no defined sequence, such as enhancers. This additional level of annotation allows for a deeper understanding of cell specific gene regulation.[14]
Clinical significance[]
Suppression of the H3K4 mono- and di-demethylase LSD-1 might extend lifespan in various species.[15]
H3K4me allows binding of MDB and increased activity of DNMT1 which could give rise to CpG island methylator phenotype (CIMP). CIMP is a type of colorectal cancers caused by the inactivation of many tumor suppressor genes from epigenetic effects.[16]
Methods[]
The histone mark H3K4me1 can be detected in a variety of ways:
1. Chromatin Immunoprecipitation Sequencing (ChIP-sequencing) measures the amount of DNA enrichment once bound to a targeted protein and immunoprecipitated. It results in good optimization and is used in vivo to reveal DNA-protein binding occurring in cells. ChIP-Seq can be used to identify and quantify various DNA fragments for different histone modifications along a genomic region.[17]
2. Micrococcal Nuclease sequencing (MNase-seq) is used to investigate regions that are bound by well positioned nucleosomes. Use of the micrococcal nuclease enzyme is employed to identify nucleosome positioning. Well positioned nucleosomes are seen to have enrichment of sequences.[18]
3. Assay for transposase accessible chromatin sequencing (ATAC-seq) is used to look in to regions that are nucleosome free (open chromatin). It uses hyperactive Tn5 transposon to highlight nucleosome localisation.[19][20][21]
See also[]
- Methamphetamine#Addiction
- Histone methylation
- Histone methyltransferase
- Methyllysine
References[]
- ^ Huang, Suming; Litt, Michael D.; Ann Blakey, C. (2015-11-30). Epigenetic Gene Expression and Regulation. pp. 21–38. ISBN 9780127999586.
- ^ Ruthenburg AJ, Li H, Patel DJ, Allis CD (December 2007). "Multivalent engagement of chromatin modifications by linked binding modules". Nature Reviews. Molecular Cell Biology. 8 (12): 983–94. doi:10.1038/nrm2298. PMC 4690530. PMID 18037899.
- ^ Kouzarides T (February 2007). "Chromatin modifications and their function". Cell. 128 (4): 693–705. doi:10.1016/j.cell.2007.02.005. PMID 17320507.
- ^ a b Rada-Iglesias, Alvaro (2018). "Is H3K4me1 at enhancers correlative or causative?". Nature Genetics. 50 (1): 4–5. doi:10.1038/s41588-017-0018-3. PMID 29273804.
- ^ Wang, Chaochen; Lee, Ji-Eun; Lai, Binbin; MacFarlan, Todd S.; Xu, Shiliyang; Zhuang, Lenan; Liu, Chengyu; Peng, Weiqun; Ge, Kai (2016). "Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition". Proceedings of the National Academy of Sciences. 113 (42): 11871–11876. doi:10.1073/pnas.1606857113. PMC 5081576. PMID 27698142.
- ^ Herz, H.-M.; Mohan, M.; Garruss, A. S.; Liang, K.; Takahashi, Y.-h.; Mickey, K.; Voets, O.; Verrijzer, C. P.; Shilatifard, A. (2012). "Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4". Genes & Development. 26 (23): 2604–2620. doi:10.1101/gad.201327.112. PMC 3521626. PMID 23166019.
- ^ a b Hyun, Kwangbeom; Jeon, Jongcheol; Park, Kihyun; Kim, Jaehoon (2017). "Writing, erasing and reading histone lysine methylations". Experimental & Molecular Medicine. 49 (4): e324. doi:10.1038/emm.2017.11. PMC 6130214. PMID 28450737.
- ^ a b c Huang, Suming; Litt, Michael D.; Ann Blakey, C. (2015-10-19). Epigenetic Gene Expression and Regulation. ISBN 9780128004715.
- ^ Jenuwein T, Allis CD (August 2001). "Translating the histone code". Science. 293 (5532): 1074–80. doi:10.1126/science.1063127. PMID 11498575.
- ^ Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. (The ENCODE Project Consortium) (June 2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820. PMID 17571346.
- ^ Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B (October 2010). "Systematic protein location mapping reveals five principal chromatin types in Drosophila cells". Cell. 143 (2): 212–24. doi:10.1016/j.cell.2010.09.009. PMC 3119929. PMID 20888037.
- ^ Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, et al. (modENCODE Consortium) (December 2010). "Identification of functional elements and regulatory circuits by Drosophila modENCODE". Science. 330 (6012): 1787–97. Bibcode:2010Sci...330.1787R. doi:10.1126/science.1198374. PMC 3192495. PMID 21177974.
- ^ Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, et al. (March 2011). "Comprehensive analysis of the chromatin landscape in Drosophila melanogaster". Nature. 471 (7339): 480–5. Bibcode:2011Natur.471..480K. doi:10.1038/nature09725. PMC 3109908. PMID 21179089.
- ^ Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, et al. (Roadmap Epigenomics Consortium) (February 2015). "Integrative analysis of 111 reference human epigenomes". Nature. 518 (7539): 317–30. Bibcode:2015Natur.518..317.. doi:10.1038/nature14248. PMC 4530010. PMID 25693563.
- ^ Sen, Payel; Shah, Parisha P.; Nativio, Raffaella; Berger, Shelley L. (2016). "Epigenetic Mechanisms of Longevity and Aging". Cell. 166 (4): 822–839. doi:10.1016/j.cell.2016.07.050. PMC 5821249. PMID 27518561.
- ^ Neidhart, Michel (2015-09-17). DNA Methylation and Complex Human Disease. p. 124. ISBN 978-0-12-420194-1.
- ^ "Whole-Genome Chromatin IP Sequencing (ChIP-Seq)" (PDF). Illumina. Retrieved 23 October 2019.
- ^ "MAINE-Seq/Mnase-Seq". illumina. Retrieved 23 October 2019.
- ^ Buenrostro, Jason D.; Wu, Beijing; Chang, Howard Y.; Greenleaf, William J. (2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109: 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. ISBN 9780471142720. PMC 4374986. PMID 25559105.
- ^ Schep, Alicia N.; Buenrostro, Jason D.; Denny, Sarah K.; Schwartz, Katja; Sherlock, Gavin; Greenleaf, William J. (2015). "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research. 25 (11): 1757–1770. doi:10.1101/gr.192294.115. ISSN 1088-9051. PMC 4617971. PMID 26314830.
- ^ Song, L.; Crawford, G. E. (2010). "DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. ISSN 1559-6095. PMC 3627383. PMID 20150147.
- Epigenetics
- Post-translational modification