Nucleic acid secondary structure

Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule.[1] The secondary structures of biological DNA's and RNA's tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.
In a non-biological context, secondary structure is a vital consideration in the nucleic acid design of nucleic acid structures for DNA nanotechnology and DNA computing, since the pattern of basepairing ultimately determines the overall structure of the molecules.
Fundamental concepts[]
Base pairing[]


In molecular biology, two nucleotides on opposite complementary DNA or RNA strands that are connected via hydrogen bonds are called a base pair (often abbreviated bp). In the canonical Watson-Crick base pairing, adenine (A) forms a base pair with thymine (T) and guanine (G) forms one with cytosine (C) in DNA. In RNA, thymine is replaced by uracil (U). Alternate hydrogen bonding patterns, such as the wobble base pair and Hoogsteen base pair, also occur—particularly in RNA—giving rise to complex and functional tertiary structures. Importantly, pairing is the mechanism by which codons on messenger RNA molecules are recognized by anticodons on transfer RNA during protein translation. Some DNA- or RNA-binding enzymes can recognize specific base pairing patterns that identify particular regulatory regions of genes. Hydrogen bonding is the chemical mechanism that underlies the base-pairing rules described above. Appropriate geometrical correspondence of hydrogen bond donors and acceptors allows only the "right" pairs to form stably. DNA with high GC-content is more stable than DNA with low GC-content, but contrary to popular belief, the hydrogen bonds do not stabilize the DNA significantly and stabilization is mainly due to stacking interactions.[2]
The larger nucleobases, adenine and guanine, are members of a class of doubly ringed chemical structures called purines; the smaller nucleobases, cytosine and thymine (and uracil), are members of a class of singly ringed chemical structures called pyrimidines. Purines are only complementary with pyrimidines: pyrimidine-pyrimidine pairings are energetically unfavorable because the molecules are too far apart for hydrogen bonding to be established; purine-purine pairings are energetically unfavorable because the molecules are too close, leading to overlap repulsion. The only other possible pairings are GT and AC; these pairings are mismatches because the pattern of hydrogen donors and acceptors do not correspond. The GU wobble base pair, with two hydrogen bonds, does occur fairly often in RNA.
Nucleic acid hybridization[]
Hybridization is the process of complementary base pairs binding to form a double helix. Melting is the process by which the interactions between the strands of the double helix are broken, separating the two nucleic acid strands. These bonds are weak, easily separated by gentle heating, enzymes, or physical force. Melting occurs preferentially at certain points in the nucleic acid.[3] T and A rich sequences are more easily melted than C and G rich regions. Particular base steps are also susceptible to DNA melting, particularly T A and T G base steps.[4] These mechanical features are reflected by the use of sequences such as TATAA at the start of many genes to assist RNA polymerase in melting the DNA for transcription.
Strand separation by gentle heating, as used in PCR, is simple providing the molecules have fewer than about 10,000 base pairs (10 kilobase pairs, or 10 kbp). The intertwining of the DNA strands makes long segments difficult to separate. The cell avoids this problem by allowing its DNA-melting enzymes (helicases) to work concurrently with topoisomerases, which can chemically cleave the phosphate backbone of one of the strands so that it can swivel around the other. Helicases unwind the strands to facilitate the advance of sequence-reading enzymes such as DNA polymerase.
Secondary structure motifs[]

Nucleic acid secondary structure is generally divided into helices (contiguous base pairs), and various kinds of loops (unpaired nucleotides surrounded by helices). Frequently these elements, or combinations of them, are further classified into additional categories including, for example, tetraloops, pseudoknots, and stem-loops.
Double helix[]
The double helix is an important tertiary structure in nucleic acid molecules which is intimately connected with the molecule's secondary structure. A double helix is formed by regions of many consecutive base pairs.
The nucleic acid double helix is a spiral polymer, usually right-handed, containing two nucleotide strands which base pair together. A single turn of the helix constitutes about ten nucleotides, and contains a major groove and minor groove, the major groove being wider than the minor groove.[5] Given the difference in widths of the major groove and minor groove, many proteins which bind to DNA do so through the wider major groove.[6] Many double-helical forms are possible; for DNA the three biologically relevant forms are A-DNA, B-DNA, and Z-DNA, while RNA double helices have structures similar to the A form of DNA.
Stem-loop structures[]
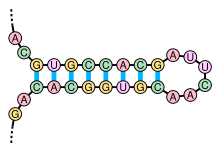
The secondary structure of nucleic acid molecules can often be uniquely decomposed into stems and loops. The stem-loop structure (also often referred to as an "hairpin"), in which a base-paired helix ends in a short unpaired loop, is extremely common and is a building block for larger structural motifs such as cloverleaf structures, which are four-helix junctions such as those found in transfer RNA. Internal loops (a short series of unpaired bases in a longer paired helix) and bulges (regions in which one strand of a helix has "extra" inserted bases with no counterparts in the opposite strand) are also frequent.
There are many secondary structure elements of functional importance to biological RNA's; some famous examples are the Rho-independent terminator stem-loops and the tRNA cloverleaf. Active research is on-going to determine the secondary structure of RNA molecules, with approaches including both experimental and computational methods (see also the List of RNA structure prediction software).
Pseudoknots[]
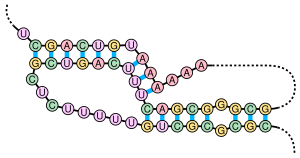
A pseudoknot is a nucleic acid secondary structure containing at least two stem-loop structures in which half of one stem is intercalated between the two halves of another stem. Pseudoknots fold into knot-shaped three-dimensional conformations but are not true topological knots. The base pairing in pseudoknots is not well nested; that is, base pairs occur that "overlap" one another in sequence position. This makes the presence of general pseudoknots in nucleic acid sequences impossible to predict by the standard method of dynamic programming, which uses a recursive scoring system to identify paired stems and consequently cannot detect non-nested base pairs with common algorithms. However, limited subclasses of pseudoknots can be predicted using modified dynamic programs.[8] Newer structure prediction techniques such as stochastic context-free grammars are also unable to consider pseudoknots.
Pseudoknots can form a variety of structures with catalytic activity[9] and several important biological processes rely on RNA molecules that form pseudoknots. For example, the RNA component of the human telomerase contains a pseudoknot that is critical for its activity.[7] The hepatitis delta virus ribozyme is a well known example of a catalytic RNA with a pseudoknot in its active site.[10][11] Though DNA can also form pseudoknots, they are generally not present in standard physiological conditions.
Secondary structure prediction[]
Most methods for nucleic acid secondary structure prediction rely on a nearest neighbor thermodynamic model.[12][13] A common method to determine the most probable structures given a sequence of nucleotides makes use of a dynamic programming algorithm that seeks to find structures with low free energy.[14] Dynamic programming algorithms often forbid pseudoknots, or other cases in which base pairs are not fully nested, as considering these structures becomes computationally very expensive for even small nucleic acid molecules. Other methods, such as stochastic context-free grammars can also be used to predict nucleic acid secondary structure.
For many RNA molecules, the secondary structure is highly important to the correct function of the RNA — often more so than the actual sequence. This fact aids in the analysis of non-coding RNA sometimes termed "RNA genes". One application of bioinformatics uses predicted RNA secondary structures in searching a genome for noncoding but functional forms of RNA. For example, microRNAs have canonical long stem-loop structures interrupted by small internal loops.
RNA secondary structure applies in RNA splicing in certain species. In humans and other tetrapods, it has been shown that without the U2AF2 protein, the splicing process is inhibited. However, in zebrafish and other teleosts the RNA splicing process can still occur on certain genes in the absence of U2AF2. This may be because 10% of genes in zebrafish have alternating TG and AC base pairs at the 3' splice site (3'ss) and 5' splice site (5'ss) respectively on each intron, which alters the secondary structure of the RNA. This suggests that secondary structure of RNA can influence splicing, potentially without the use of proteins like U2AF2 that have been thought to be required for splicing to occur.[15]
Secondary structure determination[]
RNA secondary structure can be determined from atomic coordinates (tertiary structure) obtained by X-ray crystallography, often deposited in the Protein Data Bank. Current methods include 3DNA/DSSR[16] and MC-annotate.[17]
See also[]
- DNA nanotechnology
- Molecular models of DNA
- DiProDB. The database is designed to collect and analyse thermodynamic, structural and other dinucleotide properties.
References[]
- ^ Dirks, Robert M.; Lin, Milo; Winfree, Erik & Pierce, Niles A. (2004). "Paradigms for computational nucleic acid design". Nucleic Acids Research. 32 (4): 1392–1403. doi:10.1093/nar/gkh291. PMC 390280. PMID 14990744.
- ^ Yakovchuk, Peter; Protozanova, Ekaterina; Frank-Kamenetskii, Maxim D. (2006). "Base-stacking and base-pairing contributions into thermal stability of the DNA double helix". Nucleic Acids Research. 34 (2): 564–574. doi:10.1093/nar/gkj454. PMC 1360284. PMID 16449200.
- ^ Breslauer KJ, Frank R, Blöcker H, Marky LA (1986). "Predicting DNA duplex stability from the base sequence". PNAS. 83 (11): 3746–3750. Bibcode:1986PNAS...83.3746B. doi:10.1073/pnas.83.11.3746. PMC 323600. PMID 3459152.
- ^ Richard Owczarzy (2008-08-28). "DNA melting temperature - How to calculate it?". High-throughput DNA biophysics. owczarzy.net. Retrieved 2008-10-02.
- ^ Alberts; et al. (1994). The Molecular Biology of the Cell. New York: Garland Science. ISBN 978-0-8153-4105-5.
- ^ Pabo C, Sauer R (1984). "Protein-DNA recognition". Annu Rev Biochem. 53: 293–321. doi:10.1146/annurev.bi.53.070184.001453. PMID 6236744.
- ^ a b Chen JL, Greider CW (2005). "Functional analysis of the pseudoknot structure in human telomerase RNA". Proc Natl Acad Sci USA. 102 (23): 8080–5. Bibcode:2005PNAS..102.8080C. doi:10.1073/pnas.0502259102. PMC 1149427. PMID 15849264.
- ^ Rivas E, Eddy SR (1999). "A dynamic programming algorithm for RNA structure prediction including pseudoknots". J Mol Biol. 285 (5): 2053–2068. arXiv:physics/9807048. doi:10.1006/jmbi.1998.2436. PMID 9925784. S2CID 2228845.
- ^ Staple, David W.; Butcher, Samuel E. (2005-06-14). "Pseudoknots: RNA Structures with Diverse Functions". PLOS Biol. 3 (6): e213. doi:10.1371/journal.pbio.0030213. ISSN 1545-7885. PMC 1149493. PMID 15941360.
- ^ Doudna, Jennifer A.; Ferré-D'Amaré, Adrian R.; Zhou, Kaihong (October 1998). "Crystal structure of a hepatitis delta virus ribozyme". Nature. 395 (6702): 567–574. Bibcode:1998Natur.395..567F. doi:10.1038/26912. PMID 9783582. S2CID 4359811.
- ^ Lai, Michael M. C. (1995-06-01). "The Molecular Biology of Hepatitis Delta Virus". Annual Review of Biochemistry. 64 (1): 259–286. doi:10.1146/annurev.bi.64.070195.001355. ISSN 0066-4154. PMID 7574482.
- ^ Xia T, SantaLucia J Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH (October 1998). "Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs". Biochemistry. 37 (42): 14719–35. CiteSeerX 10.1.1.579.6653. doi:10.1021/bi9809425. PMID 9778347.
- ^ Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH (May 2004). "Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure". PNAS. 101 (19): 7287–92. Bibcode:2004PNAS..101.7287M. doi:10.1073/pnas.0401799101. PMC 409911. PMID 15123812.
- ^ Zuker, M. (1989-04-07). "On finding all suboptimal foldings of an RNA molecule". Science. 244 (4900): 48–52. Bibcode:1989Sci...244...48Z. doi:10.1126/science.2468181. ISSN 0036-8075. PMID 2468181.
- ^ Lin, Chien-Ling; Taggart, Allison J.; Lim, Kian Huat; Cygan, Kamil J.; Ferraris, Luciana; Creton, Robert; Huang, Yen-Tsung; Fairbrother, William G. (13 November 2015). "RNA structure replaces the need for U2AF2 in splicing". Genome Research. 26 (1): 12–23. doi:10.1101/gr.181008.114. PMC 4691745. PMID 26566657.
- ^ Lu, XJ; Bussemaker, HJ; Olson, WK (2 December 2015). "DSSR: an integrated software tool for dissecting the spatial structure of RNA". Nucleic Acids Research. 43 (21): e142. doi:10.1093/nar/gkv716. PMC 4666379. PMID 26184874.
- ^ "MC-Annotate". www-lbit.iro.umontreal.ca.
External links[]
- MDDNA: Structural Bioinformatics of DNA
- Abalone — Commercial software for DNA modeling
- DNAlive: a web interface to compute DNA physical properties. Also allows cross-linking of the results with the UCSC Genome browser and DNA dynamics.
- DNA
- Biophysics
- Molecular geometry
- RNA