Organic photochemistry
Organic photochemistry encompasses organic reactions that are induced by the action of light.[1][2] The absorption of ultraviolet light by organic molecules often leads to reactions. In the earliest days, sunlight was employed, while in more modern times ultraviolet lamps are employed. Organic photochemistry has proven to be a very useful synthetic tool. Complex organic products can be obtained simply.
History[]
Early examples were often uncovered by the observation of precipitates or color changes from samples that were exposed to sunlights. The first reported case was by Ciamician that sunlight converted santonin to a yellow photoproduct:[3]

An early example of a precipitate was the of anthracene, characterized by Yulii Fedorovich Fritzsche and confirmed by Elbs.[4] Similar observations focused on the dimerization of cinnamic acid to truxillic acid. Many photodimers are now recognized, e.g. pyrimidine dimer, thiophosgene, diamantane.
Another example was uncovered by Egbert Havinga in 1956.[5] The curious result was activation on photolysis by a meta nitro group in contrast to the usual activation by ortho and para groups.

Organic photochemistry advanced with the development of the Woodward-Hoffmann rules.[6][7] Illustrative, these rules help rationalize the photochemically driven electrocyclic ring-closure of pent-2,4-diene, which proceeds in a disrotatory fashion.
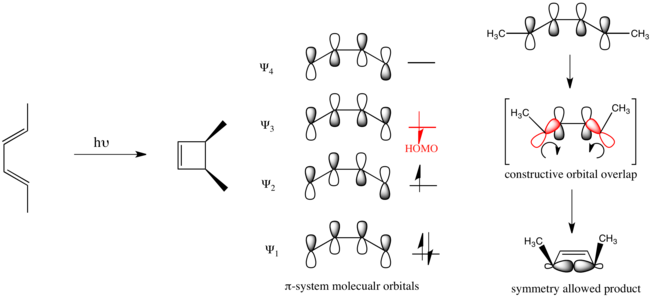
Organic reactions that obey these rules are said to be symmetry allowed. Reactions that take the opposite course are symmetry forbidden and require substantially more energy to take place if they take place at all.
Key reactions[]
Organic photochemical reactions are explained in the context of the relevant excited states.[8][9]
Parallel to the structural studies described above, the role of spin multiplicity - singlet vs triplet - on reactivity was evaluated. The importance of triplet excited species was emphasized. Triplets tend to be longer-lived than singlets and of lower energy than the singlet of the same configuration. Triplets may arise from (A) conversion of the initially formed singlets or by (B) interaction with a higher energy triplet (sensitization).
It is possible to quench a triplet reactions.[10]
Common organic photochemical reactions include: Norrish Type I, the Norrish Type II, the racemization of optically active biphenyls, the type A cyclohexadienone rearrangement, the type B cyclohexenone rearrangement, the di-pi-methane rearrangement, the type B bicyclo[3.1.0]hexanone rearrangement to phenols, photochemical electrocyclic processes, the rearrangement of epoxyketones to beta-diketones, ring opening of cyclopropyl ketones, heterolysis of 3,5-dimethoxylbenzylic derivatives, and photochemical cyclizations of dienes.
Practical considerations[]

Reactants of the photoreactions can be both gaseous and liquids.[11] In general, it is necessary to bring the reactants close to the light source in order to obtain the highest possible luminous efficacy. For this purpose, the reaction mixture can be irradiated either directly or in a flow-through side arm of a reactor with a suitable light source.[12]
A disadvantage of photochemical processes is the low efficiency of the conversion of electrical energy in the radiation energy of the required wavelength. In addition to the radiation, light sources generate plenty of heat, which in turn requires cooling energy. In addition, most light sources emit polychromatic light, even though only monochromatic light is needed.[13] A high quantum yield, however, compensates for these disadvantages.
Working at low temperatures is advantageous since side reactions are avoided (as the selectivity is increased) and the yield is increased (since gaseous reactants are driven out less from the solvent).
The starting materials can sometimes be cooled before the reaction to such an extent that the reaction heat is absorbed without further cooling of the mixture. In the case of gaseous or low-boiling starting materials, work under overpressure is necessary. Due to the large number of possible raw materials, a large number of processes have been described.[14][15] Large scale reactions are usually carried out in a stirred tank reactor, a bubble column reactor or a tube reactor, followed by further processing depending on the target product.[16] In case of a stirred tank reactor, the lamp (generally shaped as an elongated cylinder) is provided with a cooling jacket and placed in the reaction solution. Tube reactors are made from quartz or glass tubes, which are irradiated from the outside. Using a stirred tank reactor has the advantage that no light is lost to the environment. However, the intensity of light drops rapidly with the distance to the light source due to adsorption by the reactants.[12]
The influence of the radiation on the reaction rate can often be represented by a power law based on the quantum flow density, i.e. the mole light quantum (previously measured in the unit Einstein) per area and time. One objective in the design of reactors is therefore to determine the economically most favorable dimensioning with regard to an optimization of the quantum current density.[17]
Case studies[]
[2+2] Cycloadditions[]
Olefins dimerize upon UV-irradiation.[18]
4,4-Diphenylcyclohexadienone rearrangement[]
Quite parallel to the santonin to lumisantonin example is the rearrangement of 4,4-diphenylcyclohexadienone[9] Here the n-pi* triplet excited state undergoes the same beta-beta bonding. This is followed by intersystem crossing (i.e. ISC) to form the singlet ground state which is seen to be a zwitterion. The final step is the rearrangement to the bicyclic photoproduct. The reaction is termed the type A cyclohexadienone rearrangement.

4,4-diphenylcyclohexenone[]
To provide further evidence on the mechanism of the dienone in which there is bonding between the two double bonds, the case of 4,4-diphenylcyclohexenone is presented here. It is seen that the rearrangement is quite different; thus two double bonds are required for a type A rearrangement. With one double bond one of the phenyl groups, originally at C-4, has migrated to C-3 (i.e. the beta carbon).[19]

When one of the aryl groups has a para-cyano or para-methoxy group, that substituted aryl group migrates in preference.[20] Inspection of the alternative phenonium-type species, in which an aryl group has begun to migrate to the beta-carbon, reveals the greater electron delocalization with a substituent para on the migrating aryl group and thus a more stabilized pathway.

π-π* reactivity[]
Still another type of photochemical reaction is the di-pi-methane rearrangement.[21] Two further early examples were the rearrangement of 1,1,5,5-tetraphenyl-3,3-dimethyl-1,4-pentadiene (the "Mariano" molecule)[22] and the rearrangement of barrelene to semibullvalene.[23] We note that, in contrast to the cyclohexadienone reactions which used n-π* excited states, the di-π-methane rearrangements utilize π-π* excited states.

Related topics[]
Photoredox catalysis[]
In photoredox catalysis, the photon is absorbed by a sensitizer (antenna molecule or ion) which then effects redox reactions on the organic substrate. A common sensitizer is ruthenium(II) tris(bipyridine). Illustrative of photoredox catalysis are some aminotrifluoromethylation reactions.[24]

Photochlorination[]
Photochlorination is one of the largest implementations of photochemistry to organic synthesis. The photon is however not absorbed by the organic compound, but by chlorine. Photolysis of Cl2 gives chlorine atoms, which abstract H atoms from hydrocarbons, leading to chlorination.



References[]
- ^ P. Klán, J. Wirz Photochemistry of Organic Compounds: From Concepts to Practice. Wiley, Chichester, 2009, ISBN 978-1405190886.
- ^ N. J. Turro, V. Ramamurthy, J. C. Scaiano Modern Molecular Photochemistry of Organic Molecules. University Science Books, Sausalito, 2010, ISBN 978-1891389252.
- ^ Roth, Heinz D. (1989). "The Beginnings of Organic Photochemistry". Angewandte Chemie International Edition in English. 28 (9): 1193–1207. doi:10.1002/anie.198911931.
- ^ . 44. doi:10.1002/prac.18910440140. Cite journal requires
|journal=(help); Missing or empty|title=(help) - ^ Havinga, E.; De Jongh, R. O.; Dorst, W. (1956). "Photochemical acceleration of the hydrolysis of nitrophenyl phosphates and nitrophenyl sulphates". Recueil des Travaux Chimiques des Pays-Bas. 75 (4): 378–383. doi:10.1002/recl.19560750403.
- ^ Woodward, R. B.; Hoffmann, Roald (1969). "The Conservation of Orbital Symmetry". Angew. Chem. Int. Ed. 8 (11): 781–853. doi:10.1002/anie.196907811.
- ^ Woodward, R. B.; Hoffmann, Roald (1971). The Conservation of Orbital Symmetry (3rd printing, 1st ed.). Weinheim, BRD: Verlag Chemie GmbH (BRD) and Academic Press (USA). pp. 1–178. ISBN 978-1483256153.
- ^ "The Photochemical Rearrangement of 4,4-Diphenylcyclohexadienone. Paper I on a General Theory of Photochemical Reactions," Zimmerman, H. E.; Schuster, D. I. J. Am. Chem. Soc., 1961, 83, 4486-4487.
- ^ a b Zimmerman, Howard E.; David I. Schuster (1962). "A New Approach to Mechanistic Organic Photochemistry. IV. Photochemical Rearrangements of 4,4-Diphenylcyclohexadienone". Journal of the American Chemical Society. A.C.S. 84 (23): 4527–4540. doi:10.1021/ja00882a032.
- ^ "Terenin, A.; Ermolaev, V. Sensitized Phosphorescence in Organic Solutions at Low Temperature; Energy Transfer Between Triplet States", Trans. Faraday Soc., 1956, 52, 1042–1052.
- ^ Mario Schiavello (Hrsg.): Photoelectrochemistry, Photocatalysis and Photoreactors Fundamentals and Developments. Springer Netherlands, 2009, ISBN 978-90-481-8414-9, p. 564.
- ^ a b Martin Fischer: Industrial Applications of Photochemical Syntheses. In: Angewandte Chemie International Edition in English. 17, 1978, p. 16–26, doi:10.1002/anie.197800161.
- ^ Dieter Wöhrle, Michael W. Tausch, Wolf-Dieter Stohrer: Photochemie: Konzepte, Methoden, Experimente. Wiley & Sons, 1998, ISBN 978-3-527-29545-6, p. 271–275.
- ^ US Grant 1379367, F. Sparre & W. E. Masland, "Process of Chlorination", issued 1921-05-24, assigned to Du Pont
- ^ US Grant 1459777, R. Leiser & F. Ziffer, "Process and Apparatus for the Chlorination of Methane", issued 1920-02-14, assigned to Ziffer Fritz and Leiser Richard
- ^ David A. Mixon, Michael P. Bohrer, Patricia A. O’Hara: Ultrapurification of SiCl4 by photochlorination in a bubble column reactor. In: AIChE Journal. 36, 1990, p. 216–226, doi:10.1002/aic.690360207.
- ^ H. Hartig: Einfache Dimensionierung, photochemischer Reaktoren. In: Chemie Ingenieur Technik – CIT. 42, 1970, p. 1241–1245, doi:10.1002/cite.330422002.
- ^ Cargill1, R. L.; Dalton, J. R.; Morton, G. H.; Caldwell1, W. E. (1984). "Photocyclization of an Enone to an Alkene: 6-Methylbicyclo[4.2.0]Octan-2-One". Organic Syntheses. 62: 118. doi:10.15227/orgsyn.062.0118.
- ^ "Mechanistic and Exploratory Organic Photochemistry, IX. Phenyl Migration in the Irradiation of 4.4-Diphenylcyclohexenone," Zimmerman, H. E.; Wilson, J. W. J. Am. Chem. Soc., 1964, 86, 4036-4042.
- ^ "Photochemical Migratory Aptitudes in Cyclohexenones. Mechanistic and Exploratory Organic Photochemistry. XXIII," Zimmerman, H. E.; Rieke, R. D.; Scheffer, J. R. J. Am. Chem. Soc., 1967, 89, 2033-2047.
- ^ "Unsymmetrical Substitution and the Direction of the Di-pi-Methane Rearrangement; Mechanistic and Exploratory Organic Photochemistry. LVI," Zimmerman, H. E.; Pratt, A. C. J. Am. Chem. Soc., 1970, 92, 6259-6267
- ^ "The Di-pi-Methane Rearrangement. Interaction of Electronically Excited Vinyl Chromophores. Zimmerman, H. E.; Mariano, P. S. J. Am. Chem. Soc., 1969, 91, 1718-1727.
- ^ Zimmerman, H. E.; Grunewald, G. L. (1966). "The Chemistry of Barrelene. III. A Unique Photoisomerization to Semibullvalene". J. Am. Chem. Soc. 88 (1): 183–184. doi:10.1021/ja009
- ^ Yasu, Yusuke; Koike, Takashi; Akita, Munetaka (17 September 2012). "Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts". Angewandte Chemie International Edition. 51 (38): 9567–9571. doi:10.1002/anie.201205071. PMID 22936394.
- Organic chemistry
- Photochemistry