Robinson annulation
| Robinson annulation | |
|---|---|
| Named after | Robert Robinson |
| Reaction type | Ring forming reaction |
| Identifiers | |
| Organic Chemistry Portal | robinson-annulation |
| RSC ontology ID | RXNO:0000380 |
The Robinson annulation is a chemical reaction used in organic chemistry for ring formation. It was discovered by Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds.[1] The method uses a ketone and a methyl vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring by a Michael addition followed by an aldol condensation. This procedure is one of the key methods to form fused ring systems.
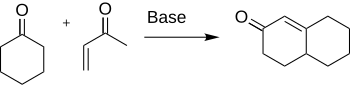
Formation of cyclohexenone and derivatives are important in chemistry for their application to the synthesis of many natural products and other interesting organic compounds such as antibiotics and steroids.[2] Specifically, the synthesis of cortisone is completed through the use of the Robinson annulation.[3]
The initial paper on the Robinson annulation was published by William Rapson and Robert Robinson while Rapson studied at Oxford with professor Robinson. Before their work, cyclohexenone syntheses were not derived from the α,β-unsaturated ketone component. Initial approaches coupled the methyl vinyl ketone with a naphthol to give a naphtholoxide, but this procedure was not sufficient to form the desired cyclohexenone. This was attributed to unsuitable conditions of the reaction.[1]
Robinson and Rapson found in 1935 that the interaction between cyclohexanone and α,β-unsaturated ketone afforded the desired cyclohexenone. It remains one of the key methods for the construction of six membered ring compounds. Since it is so widely used, there are many aspects of the reaction that have been investigated such as variations of the substrates and reaction conditions as discussed in the scope and variations section.[4] Robert Robinson won the Nobel Prize for Chemistry in 1947 for his contribution to the study of alkaloids.[5]
Reaction mechanism[]

The original procedure of the Robinson annulation begins with the nucleophilic attack of a ketone in a Michael reaction on a vinyl ketone to produce the intermediate Michael adduct. Subsequent aldol type ring closure leads to the keto alcohol, which is then followed by dehydration to produce the annulation product.
In the Michael reaction, the ketone is deprotonated by a base to form an enolate nucleophile which attacks the electron acceptor (in red). This acceptor is generally an α,β-unsaturated ketone, although aldehydes, acid derivatives and similar compounds can work as well (see scope). In the example shown here, regioselectivity is dictated by the formation of the thermodynamic enolate. Alternatively, the regioselectivity is often controlled by using a β-diketone or β-ketoester as the enolate component, since deprotonation at the carbon flanked by the carbonyl groups is strongly favored. The intramolecular aldol condensation then takes place in such a way that installs the six-membered ring. In the final product, the three carbon atoms of the α,β-unsaturated system and the carbon α to its carbonyl group make up the four-carbon bridge of the newly installed ring.
In order to avoid a reaction between the original enolate and the cyclohexenone product, the initial Michael adduct is often isolated first and then cyclized to give the desired octalone in a separate step.[6]
Stereochemistry[]
Studies have been completed on the formation of the hydroxy ketones in the Robinson annulation reaction scheme. The trans compound is favored due to antiperiplanar effects of the final aldol condensation in kinetically controlled reactions. It has also been found though that the cyclization can proceed in synclinal orientation. The figure below shows the three possible stereochemical pathways, assuming a chair transition state.[7]

It has been postulated that the difference in the formation of these transition states and their corresponding products is due to solvent interactions. Scanio found that changing the solvent of the reaction from dioxane to DMSO gives different stereochemistry in step D above. This suggests that the presence of protic or aprotic solvents gives rise to different transition states.[8]
Mechanistic classification[]

Robinson annulation is one notable example of a wider class of chemical transformations termed Tandem Michael-aldol reactions, that sequentially combine Michael addition and aldol reaction into a single reaction. As is the case with Robinson annulation, Michael addition usually happens first to tether the two reactants together, then aldol reaction proceeds intramolecularly to generate the ring system in the product. Usually five- or six-membered rings are generated.
Scope and variations[]
Reaction conditions[]
Although the Robinson annulation is generally conducted under basic conditions, reactions have been conducted under a variety of conditions. Heathcock and Ellis report similar results to the base-catalyzed method using sulfuric acid.[2] The Michael reaction can occur under neutral conditions through an enamine. A Mannich base can be heated in the presence of the ketone to produce the Michael adduct.[6] Successful preparation of compounds using the Robinson annulation methods have been reported.[9]
The Michael acceptor[]
A typical Michael acceptor is an α,β-unsaturated ketone, although aldehydes and acid derivatives work as well. In addition, Bergmann et al. reports that donors such as nitriles, nitro compounds, sulfones and certain hydrocarbons can be used as acceptors.[10] Overall, Michael acceptors are generally activated olefins such as those shown below where EWG refers to an electron withdrawing group such as cyano, keto, or ester as shown.
Wichterle reaction[]
The Wichterle reaction is a variant of the Robinson annulation that replaces methyl vinyl ketone with 1,3-dichloro-cis-2-butene. This gives an example of using a different Michael acceptor from the typical α,β-unsaturated ketone. The 1,3-dichloro-cis-2-butene is employed to avoid undesirable polymerization or condensation during the Michael addition.[11]

Hauser annulation[]
The reaction sequence in the related Hauser annulation is a Michael addition followed by a Dieckmann condensation and finally an elimination. The Dieckmann condensation is a similar ring closing intramolecular chemical reaction of diesters with base to give β-ketoesters. The Hauser donor is an aromatic sulfone or methylene sulfoxide with a carboxylic ester group in the ortho position. The Hauser acceptor is a Michael acceptor. In the original Hauser publication ethyl 2-carboxybenzyl phenyl sulfoxide reacts with pent-3-ene-2-one with LDA as a base in THF at −78 °C.[12]

Asymmetric Robinson annulation[]
Asymmetric synthesis of Robinson annulation products most often involve the use of a proline catalyst. Studies report the use of L-proline as well as several other chiral amines for use as catalysts during both steps of the Robinson annulation reaction.[13] The advantages of using the optically active proline catalysis is that they are stereoselective with enantiomeric excesses of 60–70%.[14]


Wang, et al. reported the one-pot synthesis of chiral thiochromenes by such an organocatalytic Robinson annulation.[15]
Applications to synthesis[]
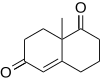
The Wieland–Miescher ketone is the Robinson annulation product of 2-methyl-cyclohexane-1,3-dione and methyl vinyl ketone. This compound is used in the syntheses of many steroids possessing important biological properties and can be made enantiopure using proline catalysis.[14]
F. Dean Toste and co-workers[16] have used Robinson annulation in the total synthesis of (+)-fawcettimine, a tetracyclic Lycopodium alkaloid that has potential application to inhibiting the acetylcholine esterase.
Enantioselective route to platensimycin[]

Scientists at Merck discovered platensimycin, a novel antibiotic lead compound with potential medicinal applications as seen in the adjacent picture.[17]
Initial synthesis gave a racemic form of the compound using an intramolecular etherification reaction of the alcohol motifs and the double bond. Yamamoto and coworkers report the use of an alternative intramolecular Robinson annulation to provide a straightforward enantioselective synthesis of tetracyclic core of platensimycin. The key Robinson annulation step was reported to be accomplished in one pot using L-proline for chiral control. The reaction conditions can be seen below.[18]

References[]
- ^ a b Rapson, William Sage; Robinson, Robert (1935). "307. Experiments on the synthesis of substances related to the sterols. Part II. A new general method for the synthesis of substituted cyclohexenones". Journal of the Chemical Society (Resumed): 1285. doi:10.1039/JR9350001285.
- ^ a b Heathcock, Clayton H.; Ellis, John E.; McMurry, John E.; Coppolino, Anthony (1971). "Acid-catalyzed Robinson Annelations". Tetrahedron Letters. 12 (52): 4995–96. doi:10.1016/s0040-4039(01)97609-9.
- ^ Acheson, R. M.; Robinson, Robert (1952). "198. Experiments bearing on the synthesis of cortisone. Part I. Some cyclopentenone derivatives". Journal of the Chemical Society (Resumed): 1127. doi:10.1039/JR9520001127.
- ^ Ho, Tse-Lok (1992). Tandem organic reactions. New York: Wiley. ISBN 978-0-471-57022-6.
- ^ McMurry, John (2008). Organic chemistry (7th ed.). Belmont, CA: Thomson Brooks/Cole. ISBN 978-0-495-11258-7.
- ^ a b Gawley, Robert E. (1976). "The Robinson Annelation and Related Reactions". Synthesis. 1976 (12): 777–794. doi:10.1055/s-1976-24200.
- ^ Nussbaumer, Cornelius (1990). "Stereochemistry of the Robinson Anellation: Studies on the Mode of Formation of the Intermediate Hydroxy Ketones". Helvetica Chimica Acta. 73 (6): 1621–1636. doi:10.1002/hlca.19900730607.
- ^ Scanio, Charles J. V.; Starrett, Richmond M. (1971). "Remarkably stereoselective Robinson annulation reaction". Journal of the American Chemical Society. 93 (6): 1539–1540. doi:10.1021/ja00735a059.
- ^ Buchschacher, Paul; A. Fürst; J. Gutzwiller (1985). "(S)-8a-Methyl-3,4,8,8a-Tetrahydro-1,6(2H, 7H)- Napthalenedione" (PDF). Organic Syntheses. 63: 37. doi:10.15227/orgsyn.063.0037. Archived from the original (PDF) on 24 April 2012.
- ^ Adams, Roger (1959). Organic Reactions. New York: John Wiley & Sons, Inc. pp. 179–555. ISBN 978-0471007593.
- ^ Wang, Zerong (2009). "Wichterle Reaction". Comprehensive organic name reactions and reagents. Hoboken, N.J.: John Wiley. doi:10.1002/9780470638859.conrr669. ISBN 978-0-470-63885-9.
- ^ Hauser, Frank M.; Rhee, Richard P. (1978). "New synthetic methods for the regioselective annelation of aromatic rings: 1-hydroxy-2,3-disubstituted naphthalenes and 1,4-dihydroxy-2,3-disubstituted naphthalenes". The Journal of Organic Chemistry. 43 (1): 178–180. doi:10.1021/jo00395a048.
- ^ Eder, Ulrich; Sauer, Gerhard; Wiechert, Rudolf (1971). "New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures". Angewandte Chemie International Edition in English. 10 (7): 496–497. doi:10.1002/anie.197104961.
- ^ a b Bui, Tommy; Barbas, Carlos F (2000). "A proline-catalyzed asymmetric Robinson annulation reaction". Tetrahedron Letters. 41 (36): 6951–6954. doi:10.1016/s0040-4039(00)01180-1.
- ^ Wang, W.; Li, H.; Wang, J.; Zu, L., J. Am. Chem. Soc. 2006; 128, 10354.
- ^ Linghu, X.; Kenedy-Smith, J. J.; Toste, F. D. (2007). "Total Synthesis of (+)-Fawcettimine". Angew. Chem. Int. Ed. 46 (40): 7671–3. doi:10.1002/anie.200702695. PMID 17729226.
- ^ Wang, Jun; Soisson, Stephen M.; Singh, Sheo; et al. (2006). "Platensimycin is a selective FabF inhibitor with potent antibiotic properties". Nature. 441 (7091): 358–61. Bibcode:2006Natur.441..358W. doi:10.1038/nature04784. PMID 16710421. S2CID 4329677. Retrieved 9 September 2021.
- ^ Li, Pingfan; Payette, Joshua N.; Yamamoto, Hisashi (2007). "Enantioselective Route to Platensimycin: An Intramolecular Robinson Annulation Approach". Journal of the American Chemical Society. 129 (31): 9534–9535. doi:10.1021/ja073547n. PMC 2553032. PMID 17630748.
- Ring forming reactions
- Name reactions
- Addition reactions
- Carbon-carbon bond forming reactions