Catalysis

Catalysis (/kəˈtæləsɪs/) is the process of increasing the rate of a chemical reaction by adding a substance known as a catalyst[1][2] (/ˈkætəlɪst/). Catalysts are not consumed in the reaction and remain unchanged after it. If the reaction is rapid and the catalyst recycles quickly, very small amounts of catalyst often suffice;[3] mixing, surface area, and temperature are important factors in reaction rate. Catalysts generally react with one or more reactants to form intermediates that subsequently give the final reaction product, in the process regenerating the catalyst.
Catalysis may be classified as either homogeneous, whose components are dispersed in the same phase (usually gaseous or liquid) as the reactant, or heterogeneous, whose components are not in the same phase. Enzymes and other biocatalysts are often considered as a third category.
Catalysis is ubiquitous in chemical industry of all kinds. Estimates are that 90% of all commercially produced chemical products involve catalysts at some stage in the process of their manufacture.
The term "catalyst" is derived from Greek καταλύειν, katalúō, meaning "loosen" or "untie". The concept of catalysis was invented by chemist Elizabeth Fulhame, based on her novel work in oxidation-reduction experiments.[4][5]
General principles[]
Illustration[]
Illustrative is the disproportionation of hydrogen peroxide to water and oxygen:
- 2 H2O2 → 2 H2O + O2
This reaction proceeds because the reaction products are more stable than the starting material. The uncatalysed reaction is slow. In fact, the decomposition of hydrogen peroxide is so slow that hydrogen peroxide solutions are commercially available. This reaction is strongly affected by catalysts such as manganese dioxide, or the enzyme peroxidase in organisms. Upon the addition of a small amount of manganese dioxide, the hydrogen peroxide reacts rapidly. This effect is readily seen by the effervescence of oxygen.[6] The manganese dioxide is not consumed in the reaction, and thus may be recovered unchanged, and re-used indefinitely. Accordingly, manganese dioxide catalyses this reaction.
Units[]
The SI derived unit for measuring the catalytic activity of a catalyst is the katal, which is quantified in moles per second. The productivity of a catalyst can be described by the turnover number (or TON) and the catalytic activity by the turn over frequency (TOF), which is the TON per time unit. The biochemical equivalent is the enzyme unit. For more information on the efficiency of enzymatic catalysis, see the article on enzymes.
Catalytic reaction mechanisms[]
In general, chemical reactions occur faster in the presence of a catalyst because the catalyst provides an alternative reaction pathway - or mechanism - with a lower activation energy than the non-catalyzed mechanism. In catalyzed mechanisms, the catalyst usually reacts to form an intermediate, which then regenerates the original catalyst in the process.[7]
Catalysts generally react with one or more reactants to form intermediates that subsequently give the final reaction product. In the process, the catalyst is regenerated. In principle, often only small amounts of catalysts are required to increase the rate of the reaction. In practice, however, catalysts are sometimes slow or are even consumed in secondary processes. The catalyst does often appear in the rate equation.[8][9][10]
As a simple example in the gas phase, the reaction 2 SO2 + O2 → 2 SO3 can be catalyzed by adding nitric oxide. The reaction mechanism occurs in two steps:
- 2 NO + O2 → 2 NO2 (slow)
- NO2 + SO2 → NO + SO3 (fast, twice)
The overall reaction is the first equation plus twice the second reaction, so that the NO catalyst is regenerated and does not participate in the net reaction. The overall rate is the rate of the slow step[10]
- v = 2k1[NO]2[O2].
As an example of a detailed mechanism for heterogeneous catalysis at a surface, when oxygen and hydrogen combine on the surface of titanium dioxide (TiO2, or titania) to produce water. Scanning tunneling microscopy showed that the molecules undergo adsorption, dissociation, and diffusion before reacting. The intermediate reaction states were: HO2, H2O2, then H3O2 and the final reaction product (water molecule dimers), after which the water molecule desorbs from the catalyst surface.[11][12]
Reaction energetics[]

Catalysts work by providing an (alternative) mechanism involving a different transition state and lower activation energy. Consequently, more molecular collisions have the energy needed to reach the transition state. Hence, catalysts can enable reactions that would otherwise be blocked or slowed by a kinetic barrier. The catalyst may increase reaction rate or selectivity, or enable the reaction at lower temperatures. This effect can be illustrated with an energy profile diagram.
In the catalyzed elementary reaction, catalysts do not change the extent of a reaction: they have no effect on the chemical equilibrium of a reaction because the rate of both the forward and the reverse reaction are both affected (see also thermodynamics).[13] The second law of thermodynamics describes why a catalyst does not change the chemical equilibrium of a reaction. Suppose there was such a catalyst that shifted an equilibrium. Introducing the catalyst to the system would result in a reaction to move to the new equilibrium, producing energy. Production of energy is a necessary result since reactions are spontaneous only if Gibbs free energy is produced, and if there is no energy barrier, there is no need for a catalyst. Then, removing the catalyst would also result in reaction, producing energy; i.e. the addition and its reverse process, removal, would both produce energy. Thus, a catalyst that could change the equilibrium would be a perpetual motion machine, a contradiction to the laws of thermodynamics.[14] Thus, catalyst does not alter the equilibrium constant. (A catalyst can however change the equilibrium concentrations by reacting in a subsequent step. It is then consumed as the reaction proceeds, and thus it is also a reactant. Illustrative is the base-catalysed hydrolysis of esters, where the produced carboxylic acid immediately reacts with the base catalyst and thus the reaction equilibrium is shifted towards hydrolysis.)
The catalyst stabilizes the transition state more than it stabilizes the starting material. It decreases the kinetic barrier by decreasing the difference in energy between starting material and transition state. It does not change the energy difference between starting materials and products (thermodynamic barrier), or the available energy (this is provided by the environment as heat or light).
Related concepts[]
Some so-called catalysts are really precatalysts. Precatalysts convert to catalysts in the reaction. For example, Wilkinson's catalyst RhCl(PPh3)3 loses one triphenylphosphine ligand before entering the true catalytic cycle. Precatalysts are easier to store but are easily activated in situ. Because of this preactivation step, many catalytic reactions involve an induction period.
Chemical species that improve catalytic activity are called co-catalysts (cocatalysts) or promoters in cooperative catalysis.
In tandem catalysis two or more different catalysts are coupled in a one-pot reaction.
In autocatalysis, the catalyst is a product of the overall reaction, in contrast to all other types of catalysis considered in this article. The simplest example of autocatalysis is a reaction of type A + B → 2 B, in one or in several steps. The overall reaction is just A → B, so that B is a product. But since B is also a reactant, it may be present in the rate equation and affect the reaction rate. As the reaction proceeds, the concentration of B increases and can accelerate the reaction as a catalyst. In effect, the reaction accelerates itself or is autocatalyzed. An example is the hydrolysis of an ester such as aspirin to a carboxylic acid and an alcohol. In the absence of added acid catalysts, the carboxylic acid product catalyzes the hydrolysis.
Classification[]
Catalysis may be classified as either homogeneous or heterogeneous. A homogeneous catalysis is one whose components are dispersed in the same phase (usually gaseous or liquid) as the reactant's molecules. A heterogeneous catalysis is one where the reaction components are not in the same phase. Enzymes and other biocatalysts are often considered as a third category. Similar mechanistic principles apply to heterogeneous, homogeneous, and biocatalysis.
Heterogeneous catalysis[]


Heterogeneous catalysts act in a different phase than the reactants. Most heterogeneous catalysts are solids that act on substrates in a liquid or gaseous reaction mixture. Important heterogeneous catalysts include zeolites, alumina,[15] higher-order oxides, graphitic carbon, transition metal oxides, metals such as Raney nickel for hydrogenation, and vanadium(V) oxide for oxidation of sulfur dioxide into sulfur trioxide by the so-called contact process.[16]
Diverse mechanisms for reactions on surfaces are known, depending on how the adsorption takes place (Langmuir-Hinshelwood, Eley-Rideal, and Mars-van Krevelen).[17] The total surface area of solid has an important effect on the reaction rate. The smaller the catalyst particle size, the larger the surface area for a given mass of particles.
A heterogeneous catalyst has active sites, which are the atoms or crystal faces where the reaction actually occurs. Depending on the mechanism, the active site may be either a planar exposed metal surface, a crystal edge with imperfect metal valence, or a complicated combination of the two. Thus, not only most of the volume but also most of the surface of a heterogeneous catalyst may be catalytically inactive. Finding out the nature of the active site requires technically challenging research. Thus, empirical research for finding out new metal combinations for catalysis continues.
For example, in the Haber process, finely divided iron serves as a catalyst for the synthesis of ammonia from nitrogen and hydrogen. The reacting gases adsorb onto active sites on the iron particles. Once physically adsorbed, the reagents undergo chemisorption that results in dissociation into adsorbed atomic species, and new bonds between the resulting fragments form in part due to their close proximity.[citation needed] In this way the particularly strong triple bond in nitrogen is broken, which would be extremely uncommon in the gas phase due to its high activation energy. Thus, the activation energy of the overall reaction is lowered, and the rate of reaction increases.[citation needed] Another place where a heterogeneous catalyst is applied is in the oxidation of sulfur dioxide on vanadium(V) oxide for the production of sulfuric acid.[16]
Heterogeneous catalysts are typically "supported," which means that the catalyst is dispersed on a second material that enhances the effectiveness or minimizes their cost. Supports prevent or reduce agglomeration and sintering small catalyst particles, exposing more surface area, thus catalysts have a higher specific activity (per gram) on a support. Sometimes the support is merely a surface on which the catalyst is spread to increase the surface area. More often, the support and the catalyst interact, affecting the catalytic reaction. Supports can also be used in nanoparticle synthesis by providing sites for individual molecules of catalyst to chemically bind. Supports are porous materials with a high surface area, most commonly alumina, zeolites or various kinds of activated carbon. Specialized supports include silicon dioxide, titanium dioxide, calcium carbonate, and barium sulfate.[citation needed]
In slurry reactions, heterogeneous catalysts can be lost by dissolving.
Many heterogeneous catalysts are in fact nanomaterials. Nanomaterial-based catalysts with enzyme-mimicking activities are collectively called as nanozymes.[18]
Electrocatalysts[]
In the context of electrochemistry, specifically in fuel cell engineering, various metal-containing catalysts are used to enhance the rates of the half reactions that comprise the fuel cell. One common type of fuel cell electrocatalyst is based upon nanoparticles of platinum that are supported on slightly larger carbon particles. When in contact with one of the electrodes in a fuel cell, this platinum increases the rate of oxygen reduction either to water, or to hydroxide or hydrogen peroxide.
Homogeneous catalysis[]
Homogeneous catalysts function in the same phase as the reactants. Typically homogeneous catalysts are dissolved in a solvent with the substrates. One example of homogeneous catalysis involves the influence of H+ on the esterification of carboxylic acids, such as the formation of methyl acetate from acetic acid and methanol.[19] High-volume processes requiring a homogeneous catalyst include hydroformylation, hydrosilylation, hydrocyanation. For inorganic chemists, homogeneous catalysis is often synonymous with organometallic catalysts.[20] Many homogeneous catalysts are however not organometallic, illustrated by the use of cobalt salts that catalyze the oxidation of p-xylene to terephthalic acid.
Organocatalysis[]
Whereas transition metals sometimes attract most of the attention in the study of catalysis, small organic molecules without metals can also exhibit catalytic properties, as is apparent from the fact that many enzymes lack transition metals. Typically, organic catalysts require a higher loading (amount of catalyst per unit amount of reactant, expressed in mol% amount of substance) than transition metal(-ion)-based catalysts, but these catalysts are usually commercially available in bulk, helping to reduce costs. In the early 2000s, these organocatalysts were considered "new generation" and are competitive to traditional metal(-ion)-containing catalysts. Organocatalysts are supposed to operate akin to metal-free enzymes utilizing, e.g., non-covalent interactions such as hydrogen bonding. The discipline organocatalysis is divided in the application of covalent (e.g., proline, DMAP) and non-covalent (e.g., thiourea organocatalysis) organocatalysts referring to the preferred catalyst-substrate binding and interaction, respectively.
Photocatalysts[]
Photocatalysis is the phenomenon where the catalyst can receive light (such as visible light), be promoted to an excited state, and then undergo intersystem crossing with the starting material, returning to ground state without being consumed. The excited state of the starting material will then undergo reactions it ordinarily could not if directly illuminated. For example, singlet oxygen is usually produced by photocatalysis. Photocatalysts are also the main ingredient in dye-sensitized solar cells.
Enzymes and biocatalysts[]
In biology, enzymes are protein-based catalysts in metabolism and catabolism. Most biocatalysts are enzymes, but other non-protein-based classes of biomolecules also exhibit catalytic properties including ribozymes, and synthetic deoxyribozymes.[21]
Biocatalysts can be thought of as intermediate between homogeneous and heterogeneous catalysts, although strictly speaking soluble enzymes are homogeneous catalysts and membrane-bound enzymes are heterogeneous. Several factors affect the activity of enzymes (and other catalysts) including temperature, pH, concentration of enzyme, substrate, and products. A particularly important reagent in enzymatic reactions is water, which is the product of many bond-forming reactions and a reactant in many bond-breaking processes.
In biocatalysis, enzymes are employed to prepare many commodity chemicals including high-fructose corn syrup and acrylamide.
Some monoclonal antibodies whose binding target is a stable molecule which resembles the transition state of a chemical reaction can function as weak catalysts for that chemical reaction by lowering its activation energy.[22] Such catalytic antibodies are sometimes called "abzymes".
Significance[]


Estimates are that 90% of all commercially produced chemical products involve catalysts at some stage in the process of their manufacture.[24] In 2005, catalytic processes generated about $900 billion in products worldwide.[25] The global demand for catalysts in 2014 was estimated at approximately US$33.5 billion.[26] Catalysis is so pervasive that subareas are not readily classified. Some areas of particular concentration are surveyed below.
Energy processing[]
Petroleum refining makes intensive use of catalysis for alkylation, catalytic cracking (breaking long-chain hydrocarbons into smaller pieces), naphtha reforming and steam reforming (conversion of hydrocarbons into synthesis gas). Even the exhaust from the burning of fossil fuels is treated via catalysis: Catalytic converters, typically composed of platinum and rhodium, break down some of the more harmful byproducts of automobile exhaust.
- 2 CO + 2 NO → 2 CO2 + N2
With regard to synthetic fuels, an old but still important process is the Fischer-Tropsch synthesis of hydrocarbons from synthesis gas, which itself is processed via water-gas shift reactions, catalysed by iron. Biodiesel and related biofuels require processing via both inorganic and biocatalysts.
Fuel cells rely on catalysts for both the anodic and cathodic reactions.
Catalytic heaters generate flameless heat from a supply of combustible fuel.
Bulk chemicals[]
Some of the largest-scale chemicals are produced via catalytic oxidation, often using oxygen. Examples include nitric acid (from ammonia), sulfuric acid (from sulfur dioxide to sulfur trioxide by the contact process), terephthalic acid from p-xylene, acrylic acid from propylene or propane and acrylonitrile from propane and ammonia.[27]
The production of ammonia is one of the largest-scale and most energy-intensive processes. In the Haber process nitrogen is combined with hydrogen over an iron oxide catalyst.[28] Methanol is prepared from carbon monoxide or carbon dioxide but using copper-zinc catalysts.
Bulk polymers derived from ethylene and propylene are often prepared via Ziegler-Natta catalysis. Polyesters, polyamides, and isocyanates are derived via acid-base catalysis.
Most carbonylation processes require metal catalysts, examples include the Monsanto acetic acid process and hydroformylation.
Fine chemicals[]
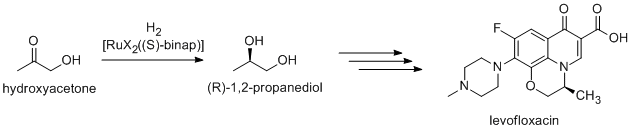
Many fine chemicals are prepared via catalysis; methods include those of heavy industry as well as more specialized processes that would be prohibitively expensive on a large scale. Examples include the Heck reaction, and Friedel–Crafts reactions. Because most bioactive compounds are chiral, many pharmaceuticals are produced by enantioselective catalysis (catalytic asymmetric synthesis).(R)-1,2-Propandiol, precursor to the antibacterial levofloxacin, can be efficiently synthesized from hydroxyacetone using Noyori asymmetric hydrogenation:[29]
Food processing[]
One of the most obvious applications of catalysis is the hydrogenation (reaction with hydrogen gas) of fats using nickel catalyst to produce margarine.[30] Many other foodstuffs are prepared via biocatalysis (see below).
Environment[]
Catalysis impacts the environment by increasing the efficiency of industrial processes, but catalysis also plays a direct role in the environment. A notable example is the catalytic role of chlorine free radicals in the breakdown of ozone. These radicals are formed by the action of ultraviolet radiation on chlorofluorocarbons (CFCs).
- Cl· + O3 → ClO· + O2
- ClO· + O· → Cl· + O2
History[]
Generally speaking,[31] anything that increases the rate of a process is a "catalyst", a term derived from Greek καταλύειν, meaning "to annul," or "to untie," or "to pick up." The concept of catalysis was invented by chemist Elizabeth Fulhame and described in a 1794 book, based on her novel work in oxidation-reduction experiments.[4][32] The first chemical reaction in organic chemistry that utilized a catalyst was studied in 1811 by Gottlieb Kirchhoff who discovered the acid-catalyzed conversion of starch to glucose. The term catalysis was later used by Jöns Jakob Berzelius in 1835[33] to describe reactions that are accelerated by substances that remain unchanged after the reaction. Fulhame, who predated Berzelius, did work with water as opposed to metals in her reduction experiments.[34] Other 18th century chemists who worked in catalysis were Eilhard Mitscherlich[35] who referred to it as contact processes, and Johann Wolfgang Döbereiner[36][37] who spoke of contact action. He developed Döbereiner's lamp, a lighter based on hydrogen and a platinum sponge, which became a commercial success in the 1820s that lives on today. Humphry Davy discovered the use of platinum in catalysis.[38] In the 1880s, Wilhelm Ostwald at Leipzig University started a systematic investigation into reactions that were catalyzed by the presence of acids and bases, and found that chemical reactions occur at finite rates and that these rates can be used to determine the strengths of acids and bases. For this work, Ostwald was awarded the 1909 Nobel Prize in Chemistry.[39] Vladimir Ipatieff performed some of the earliest industrial scale reactions, including the discovery and commercialization of oligomerization and the development of catalysts for hydrogenation.[40]
Inhibitors, poisons, and promoters[]
An added substance which does reduce the reaction rate is a reaction inhibitor if reversible and catalyst poisons if irreversible.[1] Promoters are substances that increase the catalytic activity, even though they are not catalysts by themselves.[41]
Inhibitors are sometimes referred to as "negative catalysts" since they decrease the reaction rate.[42] However the term inhibitor is preferred since they do not work by introducing a reaction path with higher activation energy; this would not reduce the rate since the reaction would continue to occur by the non-catalyzed path. Instead, they act either by deactivating catalysts, or by removing reaction intermediates such as free radicals.[42][7] In heterogeneous catalysis, coking inhibits the catalyst, which becomes covered by polymeric side products.
The inhibitor may modify selectivity in addition to rate. For instance, in the reduction of alkynes to alkenes, a palladium (Pd) catalyst partly "poisoned" with lead(II) acetate (Pb(CH3CO2)2) can be used.[43] Without the deactivation of the catalyst, the alkene produced would be further reduced to alkane.[44][45]
The inhibitor can produce this effect by, e.g., selectively poisoning only certain types of active sites. Another mechanism is the modification of surface geometry. For instance, in hydrogenation operations, large planes of metal surface function as sites of hydrogenolysis catalysis while sites catalyzing hydrogenation of unsaturates are smaller. Thus, a poison that covers the surface randomly will tend to reduce the number of uncontaminated large planes but leave proportionally more smaller sites free, thus changing the hydrogenation vs. hydrogenolysis selectivity. Many other mechanisms are also possible.
Promoters can cover up the surface to prevent production of a mat of coke, or even actively remove such material (e.g., rhenium on platinum in platforming). They can aid the dispersion of the catalytic material or bind to reagents.
See also[]
- Chemical reaction
- Abzyme
- Acid catalysis (includes Base catalysis)
- Autocatalysis
- BIG-NSE (Berlin Graduate School of Natural Sciences and Engineering)
- Catalysis Science & Technology (a chemistry journal)
- Catalytic resonance theory
- Electrocatalyst
- Environmental triggers
- Enzyme catalysis
- Industrial catalysts
- Kelvin probe force microscope
- Limiting reagent
- Pharmaceutic adjuvant
- Phase-boundary catalysis
- Phase transfer catalyst
- Photocatalysis
- Ribozyme (RNA biocatalyst)
- SUMO enzymes
- Temperature-programmed reduction
- Thermal desorption spectroscopy
References[]
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "catalyst". doi:10.1351/goldbook.C00876
- ^ Jump up to: a b "Catalyst". IUPAC Compendium of Chemical Terminology. Oxford: Blackwell Scientific Publications. 2009. doi:10.1351/goldbook.C00876. ISBN 978-0-9678550-9-7.
- ^ Masel, Richard I (2001). Chemical Kinetics and Catalysis. New York: Wiley-Interscience. ISBN 0-471-24197-0.
- ^ Lerner, Louise (2011). "7 things you may not know about catalysis". Argonne National Laboratory.
- ^ Jump up to: a b Laidler, Keith J.; Cornish-Bowden, Athel (1997). ""Elizabeth Fulhame and the discovery of catalysis: 100 years before Buchner" (PDF). In Cornish-Bowden, Athel (ed.). New beer in an old bottle : Eduard Buchner and the growth of biochemical knowledge. Valencia: Universitat de Valencia. pp. 123–126. ISBN 9788437033280. Retrieved 14 March 2021.
- ^ Rayner-Canham, Marelene; Rayner-Canham, Geoffrey William (2001). Women in Chemistry: Their Changing Roles from Alchemical Times to the Mid-Twentieth Century. American Chemical Society. ISBN 978-0-8412-3522-9.
- ^ "Genie in a Bottle". University of Minnesota. 2005-03-02. Archived from the original on 2008-04-05.
- ^ Jump up to: a b Laidler, K.J. and Meiser, J.H. (1982) Physical Chemistry, Benjamin/Cummings, p. 425. ISBN 0-618-12341-5.
- ^ Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry. Benjamin/Cummings. pp. 424–425. ISBN 0-8053-5682-7.
- ^ Atkins, Peter; de Paula, Julio (2006). Atkins' Physical Chemistry (8th ed.). W.H.Freeman. p. 839. ISBN 0-7167-8759-8.
- ^ Jump up to: a b Steinfeld, Jeffrey I.; Francisco, Joseph S.; Hase, William L. (1999). Chemical Kinetics and Dynamics (2nd ed.). Prentice Hall. pp. 147–150. ISBN 0-13-737123-3.
The catalyst concentration [C] appears in the rate expression, but not in the equilibrium ratio.
- ^ Jacoby, Mitch (16 February 2009). "Making Water Step by Step". Chemical & Engineering News. p. 10.
- ^ Matthiesen J, Wendt S, Hansen JØ, Madsen GK, Lira E, Galliker P, Vestergaard EK, Schaub R, Laegsgaard E, Hammer B, Besenbacher F (2009). "Observation of All the Intermediate Steps of a Chemical Reaction on an Oxide Surface by Scanning Tunneling Microscopy". ACS Nano. 3 (3): 517–26. CiteSeerX 10.1.1.711.974. doi:10.1021/nn8008245. ISSN 1520-605X. PMID 19309169.
- ^ Srinivasan, Bharath (2021-07-16). "A Guide to the Michaelis‐Menten equation: Steady state and beyond". The FEBS Journal. n/a (n/a): febs.16124. doi:10.1111/febs.16124. ISSN 1742-464X. PMID 34270860.
- ^ Robertson, A.J.B. (1970) Catalysis of Gas Reactions by Metals. Logos Press, London.
- ^ Shafiq, Iqrash; Shafique, Sumeer; Akhter, Parveen; Yang, Wenshu; Hussain, Murid (2020-06-23). "Recent developments in alumina supported hydrodesulfurization catalysts for the production of sulfur-free refinery products: A technical review". Catalysis Reviews. 0: 1–86. doi:10.1080/01614940.2020.1780824. ISSN 0161-4940.
- ^ Jump up to: a b Housecroft, Catherine E.; Sharpe, Alan G. (2005). Inorganic Chemistry (2nd ed.). Pearson Prentice-Hall. p. 805. ISBN 0130-39913-2.
- ^ Knözinger, Helmut and Kochloefl, Karl (2002) "Heterogeneous Catalysis and Solid Catalysts" in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim. doi:10.1002/14356007.a05_313
- ^ Wei, Hui; Wang, Erkang (2013-06-21). "Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes". Chemical Society Reviews. 42 (14): 6060–93. doi:10.1039/C3CS35486E. ISSN 1460-4744. PMID 23740388.
- ^ Behr, Arno (2002) "Organometallic Compounds and Homogeneous Catalysis" in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim. doi:10.1002/14356007.a18_215
- ^ Elschenbroich, C. (2006) Organometallics. Wiley-VCH: Weinheim. ISBN 978-3-527-29390-2
- ^ Nelson, D.L. and Cox, M.M. (2000) Lehninger, Principles of Biochemistry 3rd Ed. Worth Publishing: New York. ISBN 1-57259-153-6.
- ^ Catalytic Antibodies Simply Explained. Documentroot.com (2010-03-06). Retrieved on 2015-11-11.
- ^ Solovev, Alexander A.; Sanchez, Samuel; Mei, Yongfeng; Schmidt, Oliver G. (2011). "Tunable catalytic tubular micro-pumps operating at low concentrations of hydrogen peroxide" (PDF). Physical Chemistry Chemical Physics. 13 (21): 10131–35. Bibcode:2011PCCP...1310131S. doi:10.1039/C1CP20542K. PMID 21505711.
- ^ "Recognizing the Best in Innovation: Breakthrough Catalyst". R&D Magazine, September 2005, p. 20.
- ^ 1.4.3 Iindustrial Process Efficiency Archived 2008-05-17 at the Wayback Machine. climatetechnology.gov
- ^ "Market Report: Global Catalyst Market" (2nd ed.). Acmite Market Intelligence.
- ^ Knözinger, Helmut; Kochloefl, Karl (2003). "Heterogeneous Catalysis and Solid Catalysts". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a05_313.
- ^ Smil, Vaclav (2004). Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production (1st ed.). Cambridge, MA: MIT. ISBN 9780262693134.
- ^ Dub, Pavel A.; Gordon, John C. (2018). "The role of the metal-bound N–H functionality in Noyori-type molecular catalysts". Nature Reviews Chemistry. 2 (12): 396–408. doi:10.1038/s41570-018-0049-z. S2CID 106394152.
- ^ Clark, Jim (October 2013). "Types of catalysis". Chemguide.
- ^ Bård Lindström and Lars J. Petterson (2003) "A brief history of catalysis" Cattech, 7 (4) : 130–38.
- ^ Rayner-Canham, Marelene; Rayner-Canham, Geoffrey William (2001). Women in Chemistry: Their Changing Roles from Alchemical Times to the Mid-Twentieth Century. American Chemical Society. ISBN 978-0-8412-3522-9.
- ^ Berzelius, J.J. (1835) Årsberättelsen om framsteg i fysik och kemi [Annual report on progress in physics and chemistry]. Stockholm, Sweden: Royal Swedish Academy of Sciences. After reviewing Eilhard Mitscherlich's research on the formation of ether, Berzelius coins the word katalys (catalysis) on p. 245:
Original: Jag skall derföre, för att begagna en i kemien välkänd härledning, kalla den kroppars katalytiska kraft, sönderdelning genom denna kraft katalys, likasom vi med ordet analys beteckna åtskiljandet af kroppars beståndsdelar medelst den vanliga kemiska frändskapen.
Translation: I shall, therefore, to employ a well-known derivation in chemistry, call [the catalytic] bodies [i.e., substances] the catalytic force and the decomposition of [other] bodies by this force catalysis, just as we signify by the word analysis the separation of the constituents of bodies by the usual chemical affinities.
- ^ Srinivasan, Bharath (2020-09-27). "Words of advice: teaching enzyme kinetics". The FEBS Journal. 288 (7): 2068–2083. doi:10.1111/febs.15537. ISSN 1742-464X. PMID 32981225.
- ^ Mitscherlich, E. (1834). "Ueber die Aetherbildung" [On the formation of ether]. Annalen der Physik und Chemie. 31 (18): 273–82. Bibcode:1834AnP...107..273M. doi:10.1002/andp.18341071802.
- ^ Döbereiner (1822). "Glühendes Verbrennen des Alkohols durch verschiedene erhitzte Metalle und Metalloxyde" [Incandescent burning of alcohol by various heated metals and metal oxides]. Journal für Chemie und Physik. 34: 91–92.
- ^ Döbereiner (1823). "Neu entdeckte merkwürdige Eigenschaften des Platinsuboxyds, des oxydirten Schwefel-Platins und des metallischen Platinstaubes" [Newly discovered remarkable properties of platinum suboxide, oxidized platinum sulfide and metallic platinum dust]. Journal für Chemie und Physik. 38: 321–26.
- ^ Davy, Humphry (1817). "Some new experiments and observations on the combustion of gaseous mixtures, with an account of a method of preserving a continued light in mixtures of inflammable gases and air without flame". Philosophical Transactions of the Royal Society of London. 107: 77–85. doi:10.1098/rstl.1817.0009.
- ^ Roberts, M.W. (2000). "Birth of the catalytic concept (1800–1900)". Catalysis Letters. 67 (1): 1–4. doi:10.1023/A:1016622806065. S2CID 91507819.
- ^ Nicholas, Christopher P. (21 August 2018). "Dehydration, Dienes, High Octane, and High Pressures: Contributions from Vladimir Nikolaevich Ipatieff, a Father of Catalysis". ACS Catalysis. 8 (9): 8531–39. doi:10.1021/acscatal.8b02310.
- ^ Dhara SS; Umare SS (2018). A Textbook of Engineering Chemistry. India: S. Chand Publishing. p. 66. ISBN 9789352830688.
- ^ Jump up to: a b Laidler, K.J. (1978) Physical Chemistry with Biological Applications, Benjamin/Cummings. pp. 415–17. ISBN 0-8053-5680-0.
- ^ Lindlar, H. and Dubuis, R. (2016). "Palladium Catalyst for Partial Reduction of Acetylenes". Organic Syntheses. doi:10.15227/orgsyn.046.0089.CS1 maint: multiple names: authors list (link); Collective Volume, 5, p. 880
- ^ Jencks, W.P. (1969) Catalysis in Chemistry and Enzymology McGraw-Hill, New York. ISBN 0-07-032305-4
- ^ Bender, Myron L; Komiyama, Makoto and Bergeron, Raymond J (1984) The Bioorganic Chemistry of Enzymatic Catalysis Wiley-Interscience, Hoboken, U.S. ISBN 0-471-05991-9
External links[]
| Look up catalysis in Wiktionary, the free dictionary. |
| Wikimedia Commons has media related to Catalysis. |
| Wikisource has the text of the 1911 Encyclopædia Britannica article Catalysis. |
- Science Aid: Catalysts Page for high school level science
- W.A. Herrmann Technische Universität presentation
- Alumite Catalyst, Kameyama-Sakurai Laboratory, Japan
- Inorganic Chemistry and Catalysis Group, Utrecht University, The Netherlands
- Centre for Surface Chemistry and Catalysis
- Carbons & Catalysts Group, University of Concepcion, Chile
- Center for Enabling New Technologies Through Catalysis, An NSF Center for Chemical Innovation, USA
- "Bubbles turn on chemical catalysts", Science News magazine online, April 6, 2009.
| show Branches of chemistry |
|---|
| show Authority control |
|---|
- Catalysis
- Chemical kinetics