Barton–McCombie deoxygenation
| Barton–McCombie deoxygenation | |
|---|---|
| Named after | Derek Harold Richard Barton Stuart W. McCombie |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | barton-mccombie-reaction |
| RSC ontology ID | RXNO:0000134 |
The Barton–McCombie deoxygenation is an organic reaction in which a hydroxy functional group in an organic compound is replaced by a hydrogen to give an alkyl group.[1][2] It is named after British chemists Sir Derek Harold Richard Barton and Stuart W. McCombie.
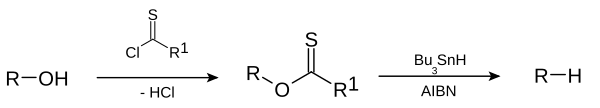
This deoxygenation reaction is a radical substitution. In the related Barton decarboxylation the reactant is a carboxylic acid.
Mechanism[]
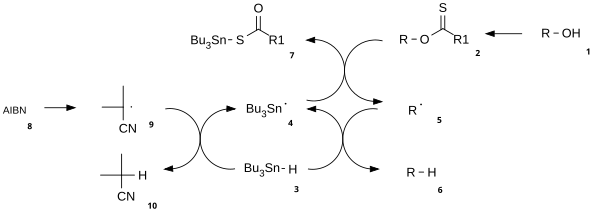
The reaction mechanism consists of a catalytic radical initiation step and a propagation step.[3] The alcohol (1) is first converted into a reactive carbonothioyl intermediate such as a thionoester or xanthate 2. Heating of AIBN results in its homolytic cleavage, generating two 2-cyanoprop-2-yl radicals 9 which each abstract a proton from tributylstannane 3 to generate tributylstannyl radicals 4 and inactive 10. The tributyltin radical abstracts the xanthate group from 2 by attack of 4 at the sulfur atom with concurrent homolytic cleavage of the C-S π bond. This leaves a carbon centered radical that forms a C-O π bond through homolytic cleavage of the R-O σ bond, giving alkyl radical 5 and tributyltin xanthate 7. The sulfur tin bond in this compound is very stable and provides the driving force for this reaction. The alkyl radical 5 then abstracts a hydrogen atom from a new molecule of tributylstannane generating the desired deoxygenated product (6) and a new radical species ready for propagation.
Variations[]
Alternative hydrogen sources[]
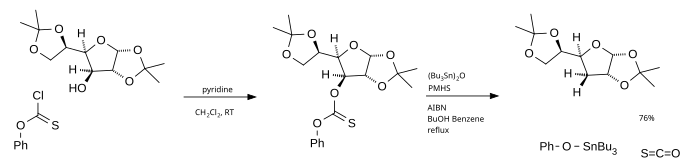
The main disadvantage of this reaction is the use of tributylstannane which is toxic, expensive and difficult to remove from the reaction mixture. One alternative is the use of tributyltin oxide as the radical source and poly(methylhydridesiloxane) (PMHS) as the hydrogen source.[4] Phenyl chlorothionoformate used as the starting material ultimately generates carbonyl sulfide.
Trialkyl boranes[]
An even more convenient hydrogen donor is provided by trialkylborane-water complexes [5] such as trimethylborane contaminated with small amounts of water.
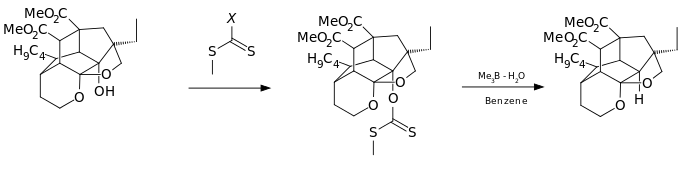
In this catalytic cycle the reaction is initiated by air oxidation of the trialkylborane 3 by air to the methyl radical 4. This radical reacts with the xanthate 2 to S-methyl-S-methyl dithiocarbonate 7 and the radical intermediate 5. The (CH3)3B.H2O complex 3 provides a hydrogen for recombining with this radical to the alkane 6 leaving behind diethyl borinic acid and a new methyl radical.
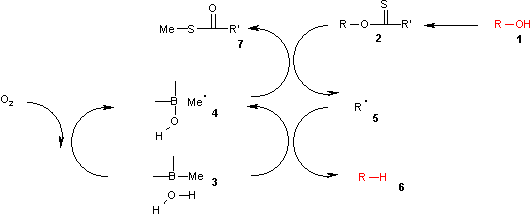
It is found by theoretical calculations that an O-H homolysis reaction in the borane-water complex is endothermic with an energy similar to that of the homolysis reaction in tributylstannane but much lower than the homolysis reaction of pure water.
Scope[]
A variation of this reaction was used as one of the steps in the total synthesis of azadirachtin:[6]

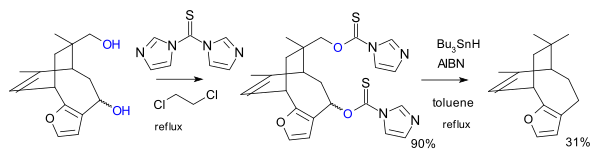
In another variation the reagent is the imidazole 1,1'-thiocarbonyldiimidazole (TCDI), for example in the total synthesis of pallescensin B.[7] TCDI is especially good to primary alcohols because there is no resonance stabilization of the xanthate because the nitrogen lonepair is involved in the aromatic sextet.[citation needed]
The reaction also applies to S-alkylxanthates. With triethylborane as a novel metal-free reagent, the required hydrogen atoms are abstracted from protic solvents, the reactor wall or even (in strictly anhydrous conditions) the borane itself.[8]
See also[]
References[]
- ^ Barton, D. H. R.; McCombie, S. W. (1975). "A new method for the deoxygenation of secondary alcohols". J. Chem. Soc., Perkin Trans. 1. 16 (16): 1574–1585. doi:10.1039/P19750001574.
- ^ Crich, D.; Quintero, L. (1989). "Radical chemistry associated with the thiocarbonyl group". Chem. Rev. 89 (7): 1413–1432. doi:10.1021/cr00097a001.
- ^ Forbes, J.E.; Zard, S.Z. (January 1989). "A novel radical chain reaction of xanthic anhydrides. Further observations on the intermediacy of alkoxy-thiocarbonyl radicals in the Barton-McCombie reaction". Tetrahedron Letters. 30 (33): 4367–4370. doi:10.1016/s0040-4039(00)99362-6.
- ^ Tormo, J.; Fu, G. C. (2002). "α-D-Ribo-hexofuranose, 3-deoxy-1,2:5,6-bis-O-(1-methylethylidene)". Org. Synth. 78: 239. doi:10.15227/orgsyn.078.0239.
- ^ Deoxygenation of Alcohols Employing Water as the Hydrogen Atom Source David A. Spiegel, Kenneth B. Wiberg, Laura N. Schacherer, Matthew R. Medeiros, and John L. Wood J. Am. Chem. Soc. 2005, 127, 12513-12515. (doi:10.1021/ja052185l)
- ^ Synthesis of Azadirachtin: A Long but Successful Journey Gemma E. Veitch, Edith Beckmann, Brenda J. Burke, Alistair Boyer, Sarah L. Maslen, and Steven V. Ley Angew. Chem. Int. Ed. 2007, doi: 10.1002/anie.200703027
- ^ The first total synthesis of (±)-pallescensin B Wen-Cheng Liu and Chun-Chen Liao ChemComm, 1999, 117–118 117 Article
- ^ Part 2. Mechanistic aspects of the reduction of S-alkyl-thionocarbonates in the presence of triethylborane and air Allais F, Boivin J, Nguyen V Beilstein J. Org. Chem., 2007 3:45 ( 12 December 2007 ) doi:10.1186/1860-5397-3-46
External links[]
- Organic redox reactions
- Free radical reactions
- Name reactions