Beta-ketothiolase deficiency
| Beta-ketothiolase deficiency | |
|---|---|
| Other names | 3-oxothiolase deficiency, Mitochondrial acetoacetyl-coenzyme A thiolase deficiency, Alpha-methyl-acetoacetyl-CoA thiolase deficiency |
 | |
| Isoleucine | |
Beta-ketothiolase deficiency is a rare, autosomal recessive metabolic disorder in which the body cannot properly process the amino acid isoleucine or the products of lipid breakdown.[1][2] Along with SCOT deficiency, it belongs to a group of disorders called ketone utilisation disorders.
The typical age of onset for this disorder is between 6 months and 24 months.
Symptoms and signs[]
The signs and symptoms of beta-ketothiolase deficiency include vomiting, dehydration, trouble breathing, extreme tiredness, and occasionally convulsions. These episodes are called ketoacidotic attacks and can sometimes lead to coma. Attacks occur when compounds called organic acids (which are formed as products of amino acid and fat breakdown) build up to toxic levels in the blood. These attacks are often triggered by an infection, fasting (not eating), or in some cases, other types of stress.[citation needed]
Genetic[]
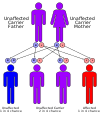
This condition is inherited in an autosomal recessive pattern and is extremely rare having only been reported in 50 to 60 individuals throughout the world.[citation needed]
Mutations in the ACAT1 gene cause beta-ketothiolase deficiency. The enzyme made by the ACAT1 gene plays an essential role in breaking down proteins and fats in the diet. Specifically, the enzyme is responsible for processing isoleucine, an amino acid that is part of many proteins. This enzyme also processes ketones, which are produced during the breakdown of fats. If a mutation in the ACAT1 gene reduces or eliminates the activity of this enzyme, the body is unable to process isoleucine and ketones properly. As a result, harmful compounds can build up and cause the blood to become too acidic (ketoacidosis), which impairs tissue function, especially in the central nervous system.[citation needed]
Diagnosis[]
In beta-ketothiolase deficiency, alpha-methyl-beta-keto-butyrate, alpha-methyl-beta-OH-butyrate and tiglyl glycine (upstream metabolites of the affected enzyme) may accumulate and may be detected on urine organic acid analysis by GC-MS. This may aid in the diagnosis, but for a more definitive diagnosis genetic confirmation needs to be done.
Treatment[]
This section is empty. You can help by . (July 2017) |
References[]
- ^ RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Beta ketothiolase deficiency". www.orpha.net. Retrieved 2017-07-02.
- ^ "OMIM Entry - # 203750 - ALPHA-METHYLACETOACETIC ACIDURIA". omim.org. Retrieved 2017-07-02.
This article incorporates public domain text from The U.S. National Library of Medicine
External links[]
| Classification | |
|---|---|
| External resources |
- Amino acid metabolism disorders
- Autosomal recessive disorders
- Rare diseases