Pulmonary hypertension
| Pulmonary hypertension | |
|---|---|
| Other names | Pulmonary arterial hypertension,[1] Ayerza syndrome[2] |
 | |
| Pulmonary hypertension | |
| Specialty | Pulmonology, cardiology |
| Symptoms | Chest pain, fatigue[3] |
| Usual onset | 20 to 60 years old[4] |
| Duration | Long term[1] |
| Causes | Unknown[1] |
| Risk factors | Family history, pulmonary embolism, HIV/AIDS, sickle cell disease, cocaine use, COPD, sleep apnea, living at high altitudes[5][4] |
| Diagnostic method | Following ruling out other potential causes[1] |
| Treatment | Supportive care, various medications, lung transplantation[1][6] |
| Medication | Epoprostenol, treprostinil, iloprost, bosentan, ambrisentan, macitentan, sildenafil[1] |
| Frequency | 1,000 new cases a year (US)[2] |
Pulmonary hypertension (PH or PHTN) is a condition of increased blood pressure within the arteries of the lungs.[7] Symptoms include shortness of breath, syncope, tiredness, chest pain, swelling of the legs, and a fast heartbeat.[7][2] The condition may make it difficult to exercise.[7] Onset is typically gradual.[8]
A patient is deemed to have pulmonary hypertension if the pulmonary mean arterial pressure is greater than 25mmHg at rest, or greater than 30mmHg during exercise.[9]
The cause is often unknown.[1] Risk factors include a family history, prior blood clots in the lungs, HIV/AIDS, sickle cell disease, cocaine use, chronic obstructive pulmonary disease, sleep apnea, living at high altitudes, and problems with the mitral valve.[5][4] The underlying mechanism typically involves inflammation and subsequent remodeling of the arteries in the lungs.[5] Diagnosis involves first ruling out other potential causes.[1]
There is currently no cure for pulmonary hypertension, although research on a cure is ongoing.[7] Treatment depends on the type of disease.[6] A number of supportive measures such as oxygen therapy, diuretics, and medications to inhibit blood clotting may be used.[1] Medications specifically used to treat pulmonary hypertension include epoprostenol, treprostinil, iloprost, bosentan, ambrisentan, macitentan, and sildenafil.[1] Lung transplantation may be an option in severe cases.[6]
The frequency of occurrence is estimated at about 1,000 new cases per year in the United States.[4][2] Females are more often affected than males.[2] Onset is typically between 20 and 60 years of age.[4] Pulmonary hypertension was identified by Ernst von Romberg in 1891.[10][1]
Classification[]
According to WHO classification there are 5 groups of PH, where Group I (pulmonary arterial hypertension) is further subdivided into Group I' and Group I'' classes.[11][12] The most recent WHO classification system (with adaptations from the more recent ESC/ERS guidelines shown in italics) can be summarized as follows:[12][13]
WHO Group I – Pulmonary arterial hypertension (PAH)
- Idiopathic
- Heritable (BMPR2, ALK1, SMAD9, caveolin 1, KCNK3 mutations)
- Drug- and toxin-induced (e.g., methamphetamine use [14] )
- Associated conditions:Connective tissue disease, HIV infection, Portal hypertension, Congenital heart diseases, Schistosomiasis
WHO Group I' – Pulmonary veno-occlusive disease (PVOD), pulmonary capillary hemangiomatosis (PCH)
- Idiopathic
- Heritable (EIF2AK4 mutations)
- Drugs, toxins and radiation-induced
- Associated conditions:connective tissue disease, HIV infection
WHO Group I" – Persistent pulmonary hypertension of the newborn
WHO Group II – Pulmonary hypertension secondary to left heart disease
- Left ventricular systolic dysfunction
- Left ventricular diastolic dysfunction
- Valvular heart disease
- Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathy
- Congenital/acquired pulmonary venous stenosis
WHO Group III – Pulmonary hypertension due to lung disease, chronic hypoxia
- Chronic obstructive pulmonary disease (COPD)
- Interstitial lung disease
- Mixed restrictive and obstructive pattern pulmonary diseases
- Sleep-disordered breathing
- Alveolar hypoventilation disorders
- Chronic exposure to high altitude
- Developmental abnormalities
WHO Group IV – chronic arterial obstruction
- Chronic thromboembolic pulmonary hypertension (CTEPH)
- Other pulmonary artery obstructions
- Angiosarcoma or other tumor within the blood vessels
- Arteritis
- Congenital pulmonary artery stenosis
- Parasitic infection (hydatidosis)
WHO Group V – Pulmonary hypertension with unclear or multifactorial mechanisms
- Hematologic diseases: chronic hemolytic anemia (including sickle cell disease)
- Systemic diseases: sarcoidosis, pulmonary Langerhans cell histiocytosis: lymphangioleiomyomatosis, neurofibromatosis, vasculitis
- Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid diseases
- Others: pulmonary tumoral thrombotic microangiopathy, fibrosing mediastinitis, chronic kidney failure, segmental pulmonary hypertension (pulmonary hypertension restricted to one or more lobes of the lungs)
Signs and symptoms[]
The symptoms of pulmonary hypertension include the following:[3][13][15]
- Shortness of breath
- Fatigue
- Chest pain
- Palpitations (heartbeat rate increased)
- Right-sided abdominal pain
- Poor appetite
- Lightheadedness
- Fainting
- Swelling (legs/ankles)
- Cyanosis
Less common signs/symptoms include non-productive cough and exercise-induced nausea and vomiting.[13] Coughing up of blood may occur in some patients, particularly those with specific subtypes of pulmonary hypertension such as heritable pulmonary arterial hypertension, Eisenmenger syndrome and chronic thromboembolic pulmonary hypertension.[16] Pulmonary venous hypertension typically presents with shortness of breath while lying flat or sleeping (orthopnea or paroxysmal nocturnal dyspnea), while pulmonary arterial hypertension (PAH) typically does not.[17]
Other typical signs of pulmonary hypertension include an accentuated pulmonary component of the second heart sound, a right ventricular third heart sound, and parasternal heave indicating a hypertrophied right ventricle. Signs of systemic congestion resulting from right-sided heart failure include jugular venous distension, ascites, and hepatojugular reflux.[13][15][18] Evidence of tricuspid insufficiency and pulmonic regurgitation is also sought and, if present, is consistent with the presence of pulmonary hypertension.[13][15][19]
Causes[]
Pulmonary hypertension is a pathophysiologic condition with many possible causes. Indeed, this condition frequently accompanies severe heart or lung conditions.[13] A 1973 World Health Organization meeting was the first attempt to classify pulmonary hypertension by its cause, and a distinction was made between primary PH (resulting from a disease of the pulmonary arteries) and secondary PH (resulting secondary to other, non-vascular causes). Further, primary PH was divided into the "arterial plexiform", "veno-occlusive" and "thromboembolic" forms.[20] In 1998, a second conference at Évian-les-Bains addressed the causes of secondary PH.[21] Subsequent third,[22] fourth,[11] and fifth (2013)[12] World Symposia on PAH have further defined the classification of PH. The classification continues to evolve based on improved understanding of the disease mechanisms.[citation needed]
Most recently in 2015, the WHO guidelines were updated by the European Society of Cardiology (ESC) and European Respiratory Society (ERS).[13] These guidelines are endorsed by the International Society for Heart and Lung Transplantation, and provide the current framework for understanding and treatment of pulmonary hypertension.[23]
Genetics[]
Mutations in several genes have been associated with this condition[24][25] these include bone morphogenetic protein receptor type 2 (BMPR2) and eukaryotic translation initiation factor 2 alpha kinase 4 gene (EIF2AK4).
Pathogenesis[]

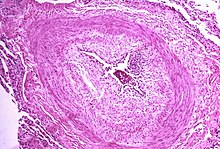
The pathogenesis of pulmonary arterial hypertension (WHO Group I) involves the narrowing of blood vessels connected to and within the lungs. This makes it harder for the heart to pump blood through the lungs, as it is much harder to make water flow through a narrow pipe as opposed to a wide one. Over time, the affected blood vessels become stiffer and thicker, in a process known as fibrosis. The mechanisms involved in this narrowing process include vasoconstriction, thrombosis, and vascular remodeling (excessive cellular proliferation, fibrosis, and reduced apoptosis/programmed cell death in the vessel walls, caused by inflammation, disordered metabolism and dysregulation of certain growth factors).[26][27] This further increases the blood pressure within the lungs and impairs their blood flow. In common with other types of pulmonary hypertension, these changes result in an increased workload for the right side of the heart.[15][28] The right ventricle is normally part of a low pressure system, with systolic ventricular pressures that are lower than those that the left ventricle normally encounters. As such, the right ventricle cannot cope as well with higher pressures, and although right ventricular adaptations (hypertrophy and increased contractility of the heart muscle) initially help to preserve stroke volume, ultimately these compensatory mechanisms are insufficient; the right ventricular muscle cannot get enough oxygen to meet its needs and right heart failure follows.[15][27][28] As the blood flowing through the lungs decreases, the left side of the heart receives less blood. This blood may also carry less oxygen than normal. Therefore, it becomes harder and harder for the left side of the heart to pump to supply sufficient oxygen to the rest of the body, especially during physical activity.[29][30][11] During the end-systolic volume phase of the cardiac cycle, the Gaussian curvature and the mean curvature of right ventricular endocardial wall of PH patients was found to be significantly different as compared to controls.[31]
In PVOD (WHO Group I'), pulmonary blood vessel narrowing occurs preferentially (though not exclusively) in post-capillary venous blood vessels.[32] PVOD shares several characteristics with PAH, but there are also some important differences, for example differences in prognosis and response to medical therapy.[citation needed]
Persistent pulmonary hypertension of the newborn occurs when the circulatory system of a newborn baby fails to adapt to life outside the womb; it is characterized by high resistance to blood flow through the lungs, right-to-left cardiac shunting and severe hypoxemia.[15]
Pathogenesis in pulmonary hypertension due to left heart disease (WHO Group II) is completely different in that constriction or damage to the pulmonary blood vessels is not the issue. Instead, the left heart fails to pump blood efficiently, leading to pooling of blood in the lungs and back pressure within the pulmonary system. This causes pulmonary edema and pleural effusions.[33] In the absence of pulmonary blood vessel narrowing, the increased back pressure is described as 'isolated post-capillary pulmonary hypertension' (older terms include 'passive' or 'proportionate' pulmonary hypertension or 'pulmonary venous hypertension'). However, in some patients, the raised pressure in the pulmonary vessels triggers a superimposed component of vessel narrowing, which further increases the workload of the right side of the heart. This is referred to as 'post-capillary pulmonary hypertension with a pre-capillary component' or 'combined post-capillary and pre-capillary pulmonary hypertension' (older terms include 'reactive' or 'out-of-proportion' pulmonary hypertension).[13][17][34]
In pulmonary hypertension due to lung diseases and/or hypoxia (WHO Group III), low levels of oxygen in the alveoli (due to respiratory disease or living at high altitude) cause constriction of the pulmonary arteries. This phenomenon is called hypoxic pulmonary vasoconstriction and it is initially a protective response designed to stop too much blood flowing to areas of the lung that are damaged and do not contain oxygen. When the alveolar hypoxia is widespread and prolonged, this hypoxia-mediated vasoconstriction occurs across a large portion of the pulmonary vascular bed and leads to an increase in pulmonary arterial pressure, with thickening of the pulmonary vessel walls contributing to the development of sustained pulmonary hypertension.[11][35][36][37] Prolonged hypoxia also induces the transcription factor HIF1A, which directly activates downstream growth factor signaling that causes irreversible proliferation and remodeling of pulmonary arterial endothelial cells, leading to chronic pulmonary arterial hypertension.[citation needed]
In CTEPH (WHO Group IV), the initiating event is thought to be blockage or narrowing of the pulmonary blood vessels with unresolved blood clots; these clots can lead to increased pressure and shear stress in the rest of the pulmonary circulation, precipitating structural changes in the vessel walls (remodeling) similar to those observed in other types of severe pulmonary hypertension. This combination of vessel occlusion and vascular remodeling once again increases the resistance to blood flow and so the pressure within the system rises.[38][39]
Molecular pathology[]
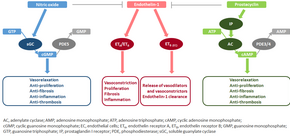
The molecular mechanism of pulmonary arterial hypertension (PAH) is not known yet, but it is believed that the endothelial dysfunction results in a decrease in the synthesis of endothelium-derived vasodilators such as nitric oxide and prostacyclin.[40] Moreover, there is a stimulation of the synthesis of vasoconstrictors such as thromboxane and vascular endothelial growth factor (VEGF). These result in a severe vasoconstriction and vascular smooth muscle and adventitial hypertrophy characteristic of patients with PAH.[40]
Nitric oxide-soluble guanylate cyclase pathway[]
In normal conditions, the vascular endothelial nitric oxide synthase produces nitric oxide from L-arginine in the presence of oxygen.[41]
This nitric oxide diffuses into neighboring cells (including vascular smooth muscle cells and platelets), where it increases the activity of the enzyme soluble guanylate cyclase, leading to increased formation of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP).[42] The cGMP then activates cGMP-dependent kinase or PKG (protein kinase G). Activated PKG promotes vasorelaxation (via a reduction of intracellular calcium levels), alters the expression of genes involved in smooth muscle cell contraction, migration and differentiation, and inhibits platelet activation.[43] Nitric oxide–soluble guanylate cyclase signaling also leads to anti-inflammatory effects.[44]
Phosphodiesterase type 5 (PDE5), which is abundant in the pulmonary tissue, hydrolyzes the cyclic bond of cGMP. Consequently, the concentration of cGMP (and thus PKG activity) decreases.[45][43]
Endothelin[]
Endothelin-1 is a peptide (comprising 21 amino acids) that is produced in endothelial cells. It acts on the endothelin receptors ETA and ETB in various cell types including vascular smooth muscle cells and fibroblasts, leading to vasoconstriction, hypertrophy, proliferation, inflammation, and fibrosis. It also acts on ETB receptors in endothelial cells; this leads to the release of both vasoconstrictors and vasodilators from those cells, and clears endothelin-1 from the system.[46][47]
Prostacyclin (and thromboxane)[]
Prostacyclin is synthesized from arachidonic acid in endothelial cells. In vascular smooth muscle cells, prostacyclin binds mainly to the prostaglandin I receptor. This sends a signal to increase adenylate cyclase activity, which leads to increased synthesis of cyclic adenosine monophosphate (cAMP). This in turn leads to increased cAMP-dependent protein kinase or PKA (protein kinase A) activity, ultimately promoting vasodilation and inhibiting cell proliferation. Prostacyclin signaling also leads to anti-thrombotic, anti-fibrotic, and anti-inflammatory effects. Levels of cAMP (which mediates most of the biological effects of prostacyclin) are reduced by phosphodiesterases 3 and 4.[48][49] The vasoconstrictor thromboxane is also synthesized from arachidonic acid. In PAH, the balance is shifted away from synthesis of prostacyclin towards synthesis of thromboxane.[48]
Other pathways[]
The three pathways described above are all targeted by currently available medical therapies for PAH. However, several other pathways have been identified that are also altered in PAH and are being investigated as potential targets for future therapies. For example, the mitochondrial enzyme pyruvate dehydrogenase kinase (PDK) is pathologically activated in PAH, causing a metabolic shift from oxidative phosphorylation to glycolysis and leading to increased cell proliferation and impaired apoptosis.[48][50] Expression of vasoactive intestinal peptide, a potent vasodilator with anti-inflammatory and immune-modulatory roles, is reduced in PAH, while expression of its receptor is increased.[48][50] Plasma levels of serotonin, which promotes vasoconstriction, hypertrophy and proliferation, are increased in patients with PAH, although the role played by serotonin in the pathogenesis of PAH remains uncertain.[15][48] The expression or activity of several growth factors (including platelet-derived growth factor, basic fibroblast growth factor, epidermal growth factor, and vascular endothelial growth factor) is increased and contributes to vascular remodeling in PAH.[48] Other factors underlying the proliferative state of pulmonary vascular smooth muscle cells include OPG[51] and TRAIL.[52] Focusing only on the pulmonary vasculature provides an incomplete picture of PAH; the ability of the right ventricle to adapt to the increased workload varies between patients and is an important determinant of survival. The molecular pathology of PAH in the right ventricle is therefore also being investigated, and recent research has shifted to consider the cardiopulmonary unit as a single system rather than two separate systems. Importantly, right ventricular remodeling is associated with increased apoptosis; this is in contrast to pulmonary vascular remodeling which involves inhibition of apoptosis.[27]
Diagnosis[]



In terms of the diagnosis of pulmonary hypertension, it has five major types, and a series of tests must be performed to distinguish pulmonary arterial hypertension from venous, hypoxic, thromboembolic, or unclear multifactorial varieties. PAH is diagnosed after exclusion of other possible causes of pulmonary hypertension.[15]
Physical examination[]
A physical examination is performed to look for typical signs of pulmonary hypertension (described above),[53] and a detailed family history is established to determine whether the disease might be heritable.[54][55][56] A history of exposure to drugs such as benfluorex (a fenfluramine derivative), dasatinib, cocaine, methamphetamine, ethanol leading to cirrhosis, and tobacco leading to emphysema is considered significant.[15][57][12] Use of selective serotonin reuptake inhibitors during pregnancy (particularly late pregnancy) is associated with an increased risk of the baby developing persistent pulmonary hypertension of the newborn.[12]
Echocardiography[]
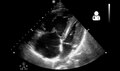
If pulmonary hypertension is suspected based on the above assessments, echocardiography is performed as the next step.[13][15][56] A meta-analysis of Doppler echocardiography for predicting the results of right heart catheterization reported a sensitivity and specificity of 88% and 56%, respectively.[58] Thus, Doppler echocardiography can suggest the presence of pulmonary hypertension, but right heart catherization (described below) remains the gold standard for diagnosis of PAH.[13][15] Echocardiography can also help to detect congenital heart disease as a cause of pulmonary hypertension.[13]
4 month old with pulmonary hypertension as seen on ultrasound[59]
4 month old with pulmonary hypertension as seen on ultrasound[59]
4 month old with pulmonary hypertension as seen on ultrasound[59]

4 month old with pulmonary hypertension as seen on ultrasound[59]
Long standing pulmonary hypertension[60]


Exclude other diseases[]
If the echocardiogram is compatible with a diagnosis of pulmonary hypertension, common causes of pulmonary hypertension (left heart disease and lung disease) are considered and further tests are performed accordingly. These tests generally include electrocardiography (ECG), pulmonary function tests including lung diffusion capacity for carbon monoxide and arterial blood gas measurements, X-rays of the chest and high-resolution computed tomography (CT) scanning.[13][15][56][61]
Ventilation/perfusion scintigraphy[]
If heart disease and lung disease have been excluded, a ventilation/perfusion scan is performed to rule out CTEPH. If unmatched perfusion defects are found, further evaluation by CT pulmonary angiography, right heart catheterization, and selective pulmonary angiography is performed.[13][56]
CT scan[]

Signs of pulmonary hypertension on CT scan of the chest are:
- Enlargement of the pulmonary trunk (measured at its bifurcation). It is, however, a poor predictor of pulmonary hypertension in patients with interstitial lung disease.[62]
- A diameter of more than 27 mm for women and 29 mm for men is suggested as a cutoff.[62]
- A cutoff of 31.6 mm may be a more statistically robust in individuals without interstitial lung disease.[62]
- Increased ratio of the diameter of the main pulmonary artery (pulmonary trunk) to the ascending aorta (measured at its bifurcation).
- Increased diameter ratio of segmental arteries to bronchi. This finding in three or four lobes, in the presence of a dilated pulmonary trunk (≥29 mm), and absence of significant structural lung disease confers a specificity of 100% for pulmonary hypertension.[62]
- Mural calcification in central pulmonary arteries is most frequently seen in patients with Eisenmenger's syndrome.[62]
Right heart catheterization[]
Although pulmonary arterial pressure (PAP) can be estimated on the basis of echocardiography,[63] pressure measurements with a Swan-Ganz catheter inserted through the right side of the heart provide the most definite assessment.[42] Pulmonary hypertension is defined as a mean PAP of at least 20 mm Hg (3300 Pa) at rest, and PAH is defined as precapillary pulmonary hypertension (i.e. mean PAP ≥ 20 mm Hg with pulmonary arterial occlusion pressure [PAOP] ≤ 15 mm Hg and pulmonary vascular resistance [PVR] > 3 Wood Units).[56] PAOP and PVR cannot be measured directly with echocardiography. Therefore, diagnosis of PAH requires right-sided cardiac catheterization. A Swan-Ganz catheter can also measure the cardiac output; this can be used to calculate the cardiac index, which is far more important in measuring disease severity than the pulmonary arterial pressure.[13][64] Mean PAP (mPAP) should not be confused with systolic PAP (sPAP), which is often reported on echocardiogram reports. A systolic pressure of 40 mm Hg typically implies a mean pressure of more than 25 mm Hg. Roughly, mPAP = 0.61•sPAP + 2.[65]
Other[]
For people considered likely to have PAH based on the above tests, the specific associated condition is then determined based on the physical examination, medical/family history and further specific diagnostic tests (for example, serological tests to detect underlying connective tissue disease, HIV infection or hepatitis, ultrasonography to confirm the presence of portal hypertension, echocardiography/cardiac magnetic resonance imaging for congenital heart disease, laboratory tests for schistosomiasis, and high-resolution CT for PVOD and pulmonary capillary hemangiomatosis). Routine lung biopsy is discouraged in patients with PAH, because of the risk to the patient and because the findings are unlikely to alter the diagnosis and treatment.[13][28][56]
Treatment[]
Treatment of pulmonary hypertension is determined by whether the PH is arterial, venous, hypoxic, thromboembolic, or miscellaneous. If it is caused by left heart disease, the treatment is to optimize left ventricular function by the use of medication or to repair/replace the mitral valve or aortic valve.[66] Patients with left heart failure or hypoxemic lung diseases (groups II or III pulmonary hypertension) should not routinely be treated with vasoactive agents including prostanoids, phosphodiesterase inhibitors, or endothelin antagonists, as these are approved for the different condition called primary pulmonary arterial hypertension.[67] To make the distinction, doctors at a minimum will conduct cardiac catheterization of the right heart, echocardiography, chest CT, a six-minute walk test, and pulmonary function testing.[67] Using treatments for other kinds of pulmonary hypertension in patients with these conditions can harm the patient and wastes substantial medical resources.[67]
High dose calcium channel blockers are useful in only 5% of IPAH patients who are vasoreactive by Swan-Ganz catheter. Unfortunately, calcium channel blockers have been largely misused, being prescribed to many patients with non-vasoreactive PAH, leading to excess morbidity and mortality.[19] The criteria for vasoreactivity have changed. Only those patients whose mean pulmonary artery pressure falls by more than 10 mm Hg to less than 40 mm Hg with an unchanged or increased cardiac output when challenged with adenosine, epoprostenol, or nitric oxide are considered vasoreactive.[68] Of these, only half of the patients are responsive to calcium channel blockers in the long term.[69]
A number of agents have recently been introduced for primary and secondary PAH. The trials supporting the use of these agents have been relatively small, and the only measure consistently used to compare their effectivity is the "6-minute walk test". Many have no data on mortality benefit or time to progression.[70]
Vasoactive substances[]
Many pathways are involved in the abnormal proliferation and contraction of the smooth muscle cells of the pulmonary arteries in patients with pulmonary arterial hypertension. Three of these pathways are important since they have been targeted with drugs — endothelin receptor antagonists, phosphodiesterase type 5 (PDE-5) inhibitors, and prostacyclin derivatives.[71]
Prostaglandins[]
Prostacyclin (prostaglandin I2) is commonly considered the most effective treatment for PAH. Epoprostenol (synthetic prostacyclin) is given via continuous infusion that requires a semi-permanent central venous catheter. This delivery system can cause sepsis and thrombosis. Prostacyclin is unstable, and therefore has to be kept on ice during administration. Since it has a half-life of 3 to 5 minutes, the infusion has to be continuous, and interruption can be fatal.[72] Other prostanoids have therefore been developed. Treprostinil can be given intravenously or subcutaneously, but the subcutaneous form can be very painful. An increased risk of sepsis with intravenous Remodulin has been reported by the CDC. Iloprost is also used in Europe intravenously and has a longer half life. Iloprost was the only inhaled form of prostacyclin approved for use in the US and Europe, until the inhaled form of treprostinil was approved by the FDA in July 2009.[medical citation needed]
Endothelin receptor antagonists[]
Moderate quality evidence suggests that endothelin receptor antagonists improve exercise capacity and decrease symptoms severity.[73] The dual (ETA and ETB) endothelin receptor antagonist bosentan was approved in 2001. Sitaxentan (Thelin) was approved for use in Canada, Australia, and the European Union,[74] but not in the United States. In 2010, Pfizer withdrew Thelin worldwide because of fatal liver complications. A similar drug, ambrisentan is marketed as Letairis in the U.S. by Gilead Sciences.[75]s
Phosphodiesterase type 5 inhibitors[]
The U.S. FDA approved sildenafil, a selective inhibitor of cGMP specific phosphodiesterase type 5 (PDE5), for the treatment of PAH in 2005. It is marketed for PAH as Revatio. In 2009, they also approved tadalafil, another PDE5 inhibitor, marketed under the name Adcirca.[76] PDE5 inhibitors are believed to increase pulmonary artery vasodilation, and inhibit vascular remodeling, thus lowering pulmonary arterial pressure and pulmonary vascular resistance.[77]
Tadalafil is taken orally, as well as sildenafil, and it is rapidly absorbed (serum levels are detectable at 20 minutes). The T1/2 (biological half-life) hovers around 17.5 hours in healthy subjects.[78] Moreover, if we consider pharmacoeconomic implications, patients that take tadalafil would pay two-thirds of the cost of sildenafil therapy.[79] However, there are some adverse effects of this drug such as headache, diarrhea, nausea, back pain, dyspepsia, flushing and myalgia.[80]
Activators of soluble guanylate cyclase[]
Soluble guanylate cyclase (sGC) is the intracellular receptor for NO. As of April 2009, the sGC activators cinaciguat and riociguat were undergoing clinical trials for the treatment of PAH.[81]
Surgical[]
Atrial septostomy is a surgical procedure that creates a communication between the right and left atria. It relieves pressure on the right side of the heart, but at the cost of lower oxygen levels in blood (hypoxia). Lung transplantation replaces a chronic condition with the ongoing need for treatment.[82] There is a post-surgical median survival of just over five years.[83]
Pulmonary thromboendarterectomy (PTE) is a surgical procedure that is used for chronic thromboembolic pulmonary hypertension. It is the surgical removal of an organized thrombus (clot) along with the lining of the pulmonary artery; it is a very difficult, major procedure that is currently performed in a few select centers.[84]
Monitoring[]
Established clinical practice guidelines dictate the frequency of pulmonary nodule evaluation and surveillance,[67][85] patients are normally monitored through commonly available tests such as:[citation needed]
- Pulse oximetry
- Arterial blood gas tests
- Chest X-rays
- Serial ECG tests
- Serial echocardiography
- Spirometry or more advanced lung function studies
- 6-minute walk test[86]
Prognosis[]

PAH is considered a universally fatal illness, although survival time may vary between individuals. The prognosis of pulmonary arterial hypertension (WHO Group I) has an untreated median survival of 2–3 years from time of diagnosis, with the cause of death usually being right ventricular failure (cor pulmonale).[87] The survival time is variable and depends on many factors.[88] A recent outcome study of those patients who had started treatment with bosentan (Tracleer) showed that 89% of patients were alive at 2 years.[89] With new therapies, survival rates are increasing. For 2,635 patients enrolled in The Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management (REVEAL Registry) from March 2006 to December 2009, 1-, 3-, 5-, and 7-year survival rates were 85%, 68%, 57%, and 49%, respectively. For patients with idiopathic/familial PAH, survival rates were 91%, 74%, 65%, and 59%.[90] Levels of mortality are very high in pregnant women with severe pulmonary arterial hypertension (WHO Group I). Pregnancy is sometimes described as contraindicated in these women.[91][92][93]
Epidemiology[]
The epidemiology of IPAH is about 125–150 deaths per year in the U.S., and worldwide the incidence is similar at 4 cases per million. However, in parts of Europe (France), indications are 6 cases per million of IPAH. Females have a higher incidence rate than males (2–9:1).[94]
Other forms of PH are far more common. In systemic scleroderma, the incidence has been estimated to be 8 to 12% of all patients;[95] in rheumatoid arthritis it is rare.[96] However, in systemic lupus erythematosus it is 4 to 14%,[97] and in sickle cell disease, it ranges from 20 to 40%.[98] Up to 4% of people who suffer a pulmonary embolism go on to develop chronic thromboembolic disease including pulmonary hypertension.[39] A small percentage of patients with COPD develop pulmonary hypertension with no other disease to explain the high pressure.[99] On the other hand, obesity-hypoventilation syndrome is very commonly associated with right heart failure due to pulmonary hypertension.[100]
Research[]
For people that inherited the disease, gene therapy is being studied.[101]
Notable cases[]
- Elaine Kaufman, American restaurateur[102]
- Ina Balin, American Broadway and TV actress[103]
- Chloe Temtchine, American singer-songwriter[104][105]
- Natalie Cole, American singer[106]
See also[]
- Pulmonary Hypertension Association
- Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR)
References[]
- ^ Jump up to: a b c d e f g h i j k "Pulmonary Arterial Hypertension – NORD (National Organization for Rare Disorders)". NORD. 2015. Archived from the original on 29 August 2017. Retrieved 30 July 2017.
- ^ Jump up to: a b c d e "Pulmonary arterial hypertension". Genetics Home Reference. January 2016. Archived from the original on 28 July 2017. Retrieved 30 July 2017.
- ^ Jump up to: a b "What Are the Signs and Symptoms of Pulmonary Hypertension? – NHLBI, NIH". www.nhlbi.nih.gov. Archived from the original on 2016-01-05. Retrieved 2015-12-30.
- ^ Jump up to: a b c d e "Who Is at Risk for Pulmonary Hypertension?". NHLBI – NIH. 2 August 2011. Archived from the original on 31 July 2017. Retrieved 30 July 2017.
- ^ Jump up to: a b c "What Causes Pulmonary Hypertension?". NHLBI – NIH. 2 August 2011. Archived from the original on 31 July 2017. Retrieved 30 July 2017.
- ^ Jump up to: a b c "How Is Pulmonary Hypertension Treated?". NHLBI – NIH. 2 August 2011. Archived from the original on 28 July 2017. Retrieved 30 July 2017.
- ^ Jump up to: a b c d "What Is Pulmonary Hypertension?". NHLBI – NIH. 2 August 2011. Archived from the original on 28 July 2017. Retrieved 30 July 2017.
- ^ "How Is Pulmonary Hypertension Diagnosed?". NHLBI – NIH. 2 August 2011. Archived from the original on 28 July 2017. Retrieved 30 July 2017.
- ^ Hatano, Shuichi; Strasser, Toma; Organization, World Health (1975). "Primary pulmonary hypertension : Report on a WHO meeting, Geneva, 15-17 October 1973". hdl:10665/39094. Cite journal requires
|journal=(help) - ^ von Romberg, Ernst (1891–1892). "Über Sklerose der Lungenarterie". Dtsch Arch Klin Med (in German). 48: 197–206.
- ^ Jump up to: a b c d Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. (June 2009). "Updated clinical classification of pulmonary hypertension". Journal of the American College of Cardiology. 54 (1 Suppl): S43–54. doi:10.1016/j.jacc.2009.04.012. PMID 19555858.
- ^ Jump up to: a b c d e Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. (December 2013). "Updated clinical classification of pulmonary hypertension". Journal of the American College of Cardiology. 62 (25 Suppl): D34–41. doi:10.1016/j.jacc.2013.10.029. PMID 24355639.
- ^ Jump up to: a b c d e f g h i j k l m n o Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. (January 2016). "2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT)". European Heart Journal. 37 (1): 67–119. doi:10.1093/eurheartj/ehv317. PMID 26320113.
- ^ Kolaitis NA, Zamanian RT, de Jesus Perez VA, Badesch DB, Benza RL, Burger CD, et al. (April 2021). "Clinical Differences and Outcomes between Methamphetamine-associated and Idiopathic Pulmonary Arterial Hypertension in the Pulmonary Hypertension Association Registry". Annals of the American Thoracic Society. 18 (4): 613–622. doi:10.1513/AnnalsATS.202007-774OC. PMC 8174020. PMID 33064950.
- ^ Jump up to: a b c d e f g h i j k l McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. (April 2009). "ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association". Journal of the American College of Cardiology. 53 (17): 1573–619. doi:10.1016/j.jacc.2009.01.004. PMID 19389575.
- ^ Diller GP, Gatzoulis MA (February 2007). "Pulmonary vascular disease in adults with congenital heart disease". Circulation. 115 (8): 1039–50. doi:10.1161/CIRCULATIONAHA.105.592386. PMID 17325254.
- ^ Jump up to: a b Fang JC, DeMarco T, Givertz MM, Borlaug BA, Lewis GD, Rame JE, et al. (September 2012). "World Health Organization Pulmonary Hypertension group 2: pulmonary hypertension due to left heart disease in the adult--a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation". The Journal of Heart and Lung Transplantation. 31 (9): 913–33. doi:10.1016/j.healun.2012.06.002. PMID 22884380.
- ^ Yusuf, Salim; Cairns, John; Camm, John; Fallen, Ernest L.; Gersh, Bernard J. (2011-09-07). Evidence-Based Cardiology. John Wiley & Sons. p. 70.3(figure). ISBN 9781444359459. Archived from the original on 2016-04-30.
- ^ Jump up to: a b "Primary Pulmonary Hypertension Clinical Presentation: History, Physical Examination, Complications". emedicine.medscape.com. Archived from the original on 2015-11-01. Retrieved 2015-12-30.
- ^ Hatano S, Strasser R (1975). Primary pulmonary hypertension. Geneva: World Health Organization.
- ^ Rich S, Rubin LJ, Abenhail L, et al. (1998). Executive summary from the World Symposium on Primary Pulmonary Hypertension (Evian, France, September 6–10, 1998). Geneva: The World Health Organization. Archived from the original on April 8, 2002.
- ^ Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. (June 2004). "Clinical classification of pulmonary hypertension". Journal of the American College of Cardiology. 43 (12 Suppl S): 5S–12S. doi:10.1016/j.jacc.2004.02.037. PMID 15194173.
- ^ Galiè, Nazzareno; Humbert, Marc; Vachiery, Jean-Luc; Gibbs, Simon; Lang, Irene; Torbicki, Adam; Simonneau, Gérald; Peacock, Andrew; Vonk Noordegraaf, Anton; Beghetti, Maurice; Ghofrani, Ardeschir; Gomez Sanchez, Miguel Angel; Hansmann, Georg; Klepetko, Walter; Lancellotti, Patrizio; Matucci, Marco; McDonagh, Theresa; Pierard, Luc A.; Trindade, Pedro T.; Zompatori, Maurizio; Hoeper, Marius (1 January 2016). "2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension". European Heart Journal. 37 (1): 67–119. doi:10.1093/eurheartj/ehv317. PMID 26320113.
- ^ Rabinovitch M (December 2012). "Molecular pathogenesis of pulmonary arterial hypertension". The Journal of Clinical Investigation. 122 (12): 4306–13. doi:10.1172/JCI60658. PMC 3533531. PMID 23202738.
- ^ Hadinnapola C, Bleda M, Haimel M, Screaton N, Swift A, Dorfmüller P, et al. (November 2017). "EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension". Circulation. 136 (21): 2022–2033. doi:10.1161/CIRCULATIONAHA.117.028351. PMC 5700414. PMID 28972005.
- ^ Jacob AS, Nielsen DH, Gianelly RE (February 1985). "Fatal ventricular fibrillation following verapamil in Wolff-Parkinson-White syndrome with atrial fibrillation". Annals of Emergency Medicine. 14 (2): 159–60. doi:10.1016/j.jacc.2013.10.025. PMC 3970402. PMID 3970402.
- ^ Jump up to: a b c Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, et al. (December 2013). "Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology". Journal of the American College of Cardiology. 62 (25 Suppl): D22–33. doi:10.1016/j.jacc.2013.10.027. PMID 24355638.
- ^ Jump up to: a b c Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. (December 2009). "Guidelines for the diagnosis and treatment of pulmonary hypertension". The European Respiratory Journal. 34 (6): 1219–63. doi:10.1183/09031936.00139009. PMID 19749199.
- ^ Yuan JX, Rubin LJ (February 2005). "Pathogenesis of pulmonary arterial hypertension: the need for multiple hits". Circulation. 111 (5): 534–8. doi:10.1161/01.CIR.0000156326.48823.55. PMID 15699271.
- ^ Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S (March 2007). "Pathology of pulmonary hypertension". Clinics in Chest Medicine. 28 (1): 23–42, vii. doi:10.1016/j.ccm.2006.11.010. PMC 1924722. PMID 17338926.
- ^ Bordones-Crom, Alifer; Patnaik, Sourav; Menon, Prahlad; Murali, Srinivas; Finol, Ender A. (2021-03-11). "Morphological Analysis of the Right Ventricular Endocardial Wall in Pulmonary Hypertension". Journal of Biomechanical Engineering. 143 (7). doi:10.1115/1.4050457. ISSN 1528-8951. PMID 33704381.
- ^ Montani D, Price LC, Dorfmuller P, Achouh L, Jaïs X, Yaïci A, Sitbon O, Musset D, Simonneau G, Humbert M (January 2009). "Pulmonary veno-occlusive disease". The European Respiratory Journal. 33 (1): 189–200. doi:10.1183/09031936.00090608. PMID 19118230.
- ^ Guazzi M, Galiè N (December 2012). "Pulmonary hypertension in left heart disease". European Respiratory Review. 21 (126): 338–46. doi:10.1183/09059180.00004612. PMID 23204122.
- ^ Vachiéry JL, Adir Y, Barberà JA, Champion H, Coghlan JG, Cottin V, et al. (December 2013). "Pulmonary hypertension due to left heart diseases". Journal of the American College of Cardiology. 62 (25 Suppl): D100–8. doi:10.1016/j.jacc.2013.10.033. PMID 24355634.
- ^ Shanks N, Macklin J, Coles S (2009). "Comparison of oral erythromycin ethylsuccinate and clavulanate-potentiated amoxicillin in the treatment of acute respiratory tract infections". Clinical Therapeutics. 11 (6): 812–9. doi:10.1016/j.coph.2009.02.006. PMC 2692823. PMID 2692823.
- ^ Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, Schulz R, Seeger W, Grimminger F, Weissmann N (December 2008). "Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms". The European Respiratory Journal. 32 (6): 1639–51. doi:10.1183/09031936.00013908. PMID 19043010.
- ^ Stenmark KR, Fagan KA, Frid MG (September 2006). "Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms". Circulation Research. 99 (7): 675–91. doi:10.1161/01.RES.0000243584.45145.3f. PMID 17008597.
- ^ McNeil K, Dunning J (September 2007). "Chronic thromboembolic pulmonary hypertension (CTEPH)". Heart. 93 (9): 1152–8. doi:10.1136/hrt.2004.053603. PMC 1955041. PMID 17699182.
- ^ Jump up to: a b Hoeper MM, Mayer E, Simonneau G, Rubin LJ (April 2006). "Chronic thromboembolic pulmonary hypertension". Circulation. 113 (16): 2011–20. doi:10.1161/CIRCULATIONAHA.105.602565. PMID 16636189.
- ^ Jump up to: a b Budhiraja R, Tuder RM, Hassoun PM (January 2004). "Endothelial dysfunction in pulmonary hypertension". Circulation. 109 (2): 159–65. doi:10.1161/01.CIR.0000102381.57477.50. PMID 14734504.
- ^ Förstermann U, Münzel T (April 2006). "Endothelial nitric oxide synthase in vascular disease: from marvel to menace". Circulation. 113 (13): 1708–14. doi:10.1161/CIRCULATIONAHA.105.602532. PMID 16585403.
- ^ Murad F (November 2006). "Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development". The New England Journal of Medicine. 355 (19): 2003–11. doi:10.1056/NEJMsa063904. PMID 17093251.
- ^ Jump up to: a b Francis SH, Busch JL, Corbin JD, Sibley D (2010). "cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action". Pharmacol Rev. 62 (3): 525–63. doi:10.1124/pr.110.002907. PMC 2964902. PMID 20716671.CS1 maint: multiple names: authors list (link)
- ^ Zelia OP, Kovalenko FP (2011). "[Comparative efficiency of infecting laboratory animals via intravenous and subcutaneous administration of Schistosoma mansoni cercaria]". Parazitologiia. 20 (6): 461–5. PMID 3103045.
- ^ Ghofrani HA, Pepke-Zaba J, Barbera JA, Channick R, Keogh AM, Gomez-Sanchez MA, et al. (June 2004). "Nitric oxide pathway and phosphodiesterase inhibitors in pulmonary arterial hypertension". Journal of the American College of Cardiology. 43 (12 Suppl S): 68S–72S. doi:10.1016/j.jacc.2004.02.031. PMID 15194181.
- ^ McLaughlin VV, McGoon MD (September 2006). "Pulmonary arterial hypertension". Circulation. 114 (13): 1417–31. doi:10.1161/CIRCULATIONAHA.104.503540. PMID 17000921.
- ^ Fonseca C, Abraham D, Renzoni EA (January 2011). "Endothelin in pulmonary fibrosis". American Journal of Respiratory Cell and Molecular Biology. 44 (1): 1–10. doi:10.1165/rcmb.2009-0388TR. PMID 20448055.
- ^ Jump up to: a b c d e f Wood SF (January 1986). "Astemizole and terfenadine compared in hay fever". The Practitioner. 230 (1411): 41–4. doi:10.1161/CIRCULATIONAHA.108.847707. PMID 2869481.
- ^ Gomberg-Maitland M, Olschewski H (April 2008). "Prostacyclin therapies for the treatment of pulmonary arterial hypertension". The European Respiratory Journal. 31 (4): 891–901. doi:10.1183/09031936.00097107. PMID 18378784.
- ^ Jump up to: a b Lenfant M (2013). "[Dihydrostreptomycin fixation on the ribosomes of E. coli]". Biochimie. 54 (2): 283–5. doi:10.1016/j.jacc.2013.10.026. PMID 4117578.
- ^ Lawrie, Allan (2014). "The role of the osteoprotegerin/tumor necrosis factor related apoptosis-inducing ligand axis in the pathogenesis of pulmonary arterial hypertension". Vascular Pharmacology. 63 (3): 114–117. doi:10.1016/j.vph.2014.10.002. ISSN 1537-1891. PMID 25446166.
- ^ Braithwaite, Adam T.; Marriott, Helen M.; Lawrie, Allan (2018). "Divergent Roles for TRAIL in Lung Diseases". Frontiers in Medicine. 5: 212. doi:10.3389/fmed.2018.00212. ISSN 2296-858X. PMC 6072839. PMID 30101145.
- ^ "How Is Pulmonary Hypertension Diagnosed? – NHLBI, NIH". www.nhlbi.nih.gov. Archived from the original on 2016-01-05. Retrieved 2015-12-30.
- ^ "Pulmonary arterial hypertension". Genetics Home Reference. 2015-12-28. Archived from the original on 2015-12-24. Retrieved 2015-12-30.
- ^ Austin, Eric D.; Loyd, James E.; Phillips, John A. (1993-01-01). "Heritable Pulmonary Arterial Hypertension Overview". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora J.H.; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Heritable Pulmonary Arterial Hypertension. Seattle (WA): University of Washington, Seattle. PMID 20301658. Archived from the original on 2017-09-10.|Online 2015
- ^ Jump up to: a b c d e f Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. (December 2013). "Definitions and diagnosis of pulmonary hypertension". Journal of the American College of Cardiology. 62 (25 Suppl): D42–50. doi:10.1016/j.jacc.2013.10.032. PMID 24355641.
- ^ Klepper, Michael J.; Cobert, Barton (2010-10-25). Drug Safety Data: How to Analyze, Summarize and Interpret to Determine Risk. Jones & Bartlett Learning. p. 86. ISBN 9780763769123. Archived from the original on 2016-05-04.
- ^ Taleb M, Khuder S, Tinkel J, Khouri SJ (March 2013). "The diagnostic accuracy of Doppler echocardiography in assessment of pulmonary artery systolic pressure: a meta-analysis". Echocardiography. 30 (3): 258–65. doi:10.1111/echo.12061. PMID 23227919. S2CID 7460778.
- ^ Jump up to: a b c d "UOTW #29 - Ultrasound of the Week". Ultrasound of the Week. 16 December 2014. Archived from the original on 9 May 2017. Retrieved 27 May 2017.
- ^ "UOTW #65 - Ultrasound of the Week". Ultrasound of the Week. 20 December 2015. Archived from the original on 3 November 2016. Retrieved 27 May 2017.
- ^ "How Is Pulmonary Hypertension Diagnosed?". National Heart, Lung, and Blood Institute.
- ^ Jump up to: a b c d e f g Frank Gaillard. "Pulmonary hypertension". Radiopaedia. Retrieved 2018-03-12.
- ^ Bossone E, D'Andrea A, D'Alto M, Citro R, Argiento P, Ferrara F, et al. (January 2013). "Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis". Journal of the American Society of Echocardiography. 26 (1): 1–14. doi:10.1016/j.echo.2012.10.009. PMID 23140849.
- ^ Swan-Ganz – right heart catheterization: MedlinePlus Medical Encyclopedia. Accessed: 2015-12-30
- ^ Khouri SJ, Pandya U (2012). Pulmonary hypertension. In: Garcia MJ, editor. NonInvasive cardiovascular imaging: a multimodality approach: Lippincott Williams & Wilkins. p. 655-68
- ^ "How Is Pulmonary Hypertension Treated? – NHLBI, NIH". www.nhlbi.nih.gov. Archived from the original on 2016-01-05. Retrieved 2015-12-30.
- ^ Jump up to: a b c d American College of Chest Physicians; American Thoracic Society (September 2013), "Five Things Physicians and Patients Should Question", Choosing Wisely: an initiative of the ABIM Foundation, American College of Chest Physicians and American Thoracic Society, archived from the original on 3 November 2013, retrieved 6 January 2013, which cites
- McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. (April 2009). "ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association". Circulation. 119 (16): 2250–94. doi:10.1161/CIRCULATIONAHA.109.192230. PMID 19332472.
- ESC Committee for Practice Guidelines (CPG), Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, et al. (October 2009). "Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT)". European Heart Journal. 30 (20): 2493–537. doi:10.1093/eurheartj/ehp297. PMID 19713419.
- Hoeper MM, Barberà JA, Channick RN, Hassoun PM, Lang IM, Manes A, et al. (June 2009). "Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension". Journal of the American College of Cardiology. 54 (1 Suppl): S85–96. doi:10.1016/j.jacc.2009.04.008. PMID 19555862.
- ^ Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, Gaine S (June 2004). "Diagnosis and differential assessment of pulmonary arterial hypertension". Journal of the American College of Cardiology. 43 (12 Suppl S): 40S–47S. doi:10.1016/j.jacc.2004.02.032. PMID 15194177.
- ^ Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, et al. (June 2005). "Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension". Circulation. 111 (23): 3105–11. doi:10.1161/CIRCULATIONAHA.104.488486. PMID 15939821.
- ^ Torres F (October 2007). "Systematic review of randomised, double-blind clinical trials of oral agents conducted in patients with pulmonary arterial hypertension". International Journal of Clinical Practice. 61 (10): 1756–65. doi:10.1111/j.1742-1241.2007.01545.x. PMID 17877662. S2CID 1191543.
- ^ Raja SG, Raja SM (November 2011). "Treating pulmonary arterial hypertension: current treatments and future prospects". Therapeutic Advances in Chronic Disease. 2 (6): 359–70. doi:10.1177/2040622311420773. PMC 3513893. PMID 23251761.
- ^ Safdar, Zeenat (2011). "Treatment of pulmonary arterial hypertension: The role of prostacyclin and prostaglandin analogs – Respiratory Medicine". Respiratory Medicine. 105 (6): 818–827. doi:10.1016/j.rmed.2010.12.018. PMID 21273054. Retrieved 2015-12-30.
- ^ Liu, Chao; Chen, Junmin; Gao, Yanqiu; Deng, Bao; Liu, Kunshen (2021-03-25). Cochrane Airways Group (ed.). "Endothelin receptor antagonists for pulmonary arterial hypertension". Cochrane Database of Systematic Reviews. 2021 (3): CD004434. doi:10.1002/14651858.CD004434.pub6. PMC 8094512. PMID 33765691.
- ^ "UPDATE 1-Encysive gets Canadian approval for hypertension drug". Reuters. 2008-05-30. Archived from the original on 2007-07-04. Retrieved 2007-07-08.
- ^ "U.S. Food and Drug Administration Approves Gilead's Letairis Treatment of Pulmonary Arterial Hypertension" (Press release). Gilead Sciences. 2007-06-15. Archived from the original on 2007-09-27. Retrieved 2007-06-16.
- ^ "FDA approves Adcirca (tadalafil) tablets for pulmonary arterial hypertension" (Press release). 2009-05-26. Archived from the original on 2010-12-03. Retrieved 2010-12-06.
- ^ Duarte JD, Hanson RL, Machado RF (May 2013). "Pharmacologic treatments for pulmonary hypertension: exploring pharmacogenomics". Future Cardiology. 9 (3): 335–49. doi:10.2217/fca.13.6. PMC 3864092. PMID 23668740.
- ^ Forgue ST, Patterson BE, Bedding AW, Payne CD, Phillips DL, Wrishko RE, Mitchell MI (March 2006). "Tadalafil pharmacokinetics in healthy subjects". British Journal of Clinical Pharmacology. 61 (3): 280–8. doi:10.1111/j.1365-2125.2005.02553.x. PMC 1885023. PMID 16487221.
- ^ Sally A. Arif, PharmD, BCPS (Department of Pharmacy Practice, Chicago College of Pharmacy, Midwestern University, Downers Grove, Illinois, and Department of Pharmacy, Rush University Medical Center, Chicago, Illinois); and Henry Poon, PharmD, BCPS (Department of Pharmacy, James J. Peters VA Medical Center, Bronx, New York). Tadalafil: A Long-Acting Phosphodiesterase-5 Inhibitor for the Treatment of Pulmonary Arterial Hypertension. 2011;33:993–1004
- ^ Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. (June 2009). "Tadalafil therapy for pulmonary arterial hypertension". Circulation. 119 (22): 2894–903. doi:10.1161/circulationaha.108.839274. PMID 19470885.
- ^ Lasker GF, Maley JH, Pankey EA, Kadowitz PJ (April 2011). "Targeting soluble guanylate cyclase for the treatment of pulmonary hypertension". Expert Review of Respiratory Medicine. 5 (2): 153–61. doi:10.1586/ers.11.9. PMC 3108035. PMID 21510726.
- ^ George, MP; Champion, HC; Pilewski, JM (2011). "Lung transplantation for pulmonary hypertension". Pulmonary Circulation. 1 (2): 182–91. doi:10.4103/2045-8932.83455. PMC 3198646. PMID 22034605.
- ^ "2006 OPTN/SRTR Annual Report". US Scientific Registry of Transplant Recipients. 2006-05-01. Archived from the original on 2010-06-05. Retrieved 2007-03-28.
- ^ Cerveri I, D'Armini AM, Viganò M (April 2003). "Pulmonary thromboendarterectomy almost 50 years after the first surgical attempts". Heart. 89 (4): 369–70. doi:10.1136/heart.89.4.369. PMC 1769265. PMID 12639858.
- ^ American College of Chest Physicians; American Thoracic Society (September 2013), "Five Things Physicians and Patients Should Question", Choosing Wisely: an initiative of the ABIM Foundation, American College of Chest Physicians and American Thoracic Society, archived from the original on 3 November 2013, retrieved 6 January 2013
- ^ Guidelines: Six-minute Walk Test. Accessed: 2015
- ^ "Pulmonary Hypertension. About Pulmonary Hypertension | Patient". Patient. Archived from the original on 2016-01-02. Retrieved 2015-12-30.
- ^ Mouratoglou, Sophia Anastasia; Mayoumy, Ahmed A; Noordegraaf, Anton Vonk (2021). "Prediction Models and Scores in Pulmonary Hypertension: A Review". Current Pharmaceutical Design. 27 (10): 1266–1276. doi:10.2174/1381612824999201105163437. PMID 33155897.
- ^ McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galiè N, et al. (February 2005). "Survival with first-line bosentan in patients with primary pulmonary hypertension". The European Respiratory Journal. 25 (2): 244–9. doi:10.1183/09031936.05.00054804. PMID 15684287.
- ^ Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD (August 2012). "An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry". Chest. 142 (2): 448–456. doi:10.1378/chest.11-1460. PMID 22281797. [FREE]
- ^ Kaufman, Matthew H.; Latha Stead; Feig, Robert (2007). First aid for the obstetrics & gynecology clerkship. New York: McGraw-Hill, Medical Pub. Division. p. 100. ISBN 978-0-07-144874-1.
- ^ Ghosh, Amit K. (2008). Mayo Clinic Internal Medicine Review: Eighth Edition (Mayo Clinic Internal Medicine Review). Informa Healthcare. p. 55. ISBN 978-1-4200-8478-8.
- ^ British Journal of Anaesthesia: "Primary pulmonary hypertension in pregnancy; a role for novel vasodilators" Archived 2011-01-21 at the Wayback Machine March 19, 2011
- ^ "Primary Pulmonary Hypertension: Practice Essentials, Background, Pathophysiology". 2018-11-30. Archived from the original on 2015-12-24. Cite journal requires
|journal=(help) - ^ York M, Farber HW (November 2011). "Pulmonary hypertension: screening and evaluation in scleroderma". Current Opinion in Rheumatology. 23 (6): 536–44. doi:10.1097/BOR.0b013e32834ba6a7. PMID 21934501. S2CID 19528958.
- ^ Nannini, Carlotta. "Lung Disease in Rheumatoid Arthritis". MedScape.com. MedScape. Archived from the original on 8 February 2016. Retrieved 31 December 2015.
- ^ Mittoo, Shikha (2014). "Pulmonary Manifestations of Systemic Lupus Erythematosus". Seminars in Respiratory and Critical Care Medicine. MedScape. 35 (2): 249–54. doi:10.1055/s-0034-1371537. PMID 24668539. Archived from the original on 26 October 2015. Retrieved 31 December 2015.
- ^ Lee MT, Rosenzweig EB, Cairo MS (August 2007). "Pulmonary hypertension in sickle cell disease". Clinical Advances in Hematology & Oncology. 5 (8): 645–53, 585. PMID 17982405.
- ^ Minai OA, Chaouat A, Adnot S (June 2010). "Pulmonary hypertension in COPD: epidemiology, significance, and management: pulmonary vascular disease: the global perspective". Chest. 137 (6 Suppl): 39S–51S. doi:10.1378/chest.10-0087. PMID 20522579.
- ^ Balachandran JS, Masa JF, Mokhlesi B (September 2014). "Obesity Hypoventilation Syndrome Epidemiology and Diagnosis". Sleep Medicine Clinics. 9 (3): 341–347. doi:10.1016/j.jsmc.2014.05.007. PMC 4210766. PMID 25360072.
- ^ Reynolds, PN (February 2011). "Gene therapy for pulmonary hypertension: prospects and challenges". Expert Opinion on Biological Therapy. 11 (2): 133–43. doi:10.1517/14712598.2011.542139. PMID 21219232. S2CID 23641857.
- ^ "Elaine Kaufman, famed Elaine's restaurateur, dies at age 81". Daily News. Archived from the original on 28 August 2016. Retrieved 27 September 2016.
- ^ FOLKART, BURT A. (21 June 1990). "Ina Balin, 52; Movie and TV Actress Sought Lung Implant". Los Angeles Times. Archived from the original on 11 September 2016. Retrieved 27 September 2016.
- ^ Phull, Hardeep (July 26, 2016). "She's tethered to an oxygen tank, but her singing career is soaring". New York Post. Archived from the original on August 14, 2016.
- ^ Insdorf, Annette (November 10, 2013). "The Challenges of Chloe Temtchine". The Huffington Post. Archived from the original on September 18, 2016.
- ^ "Autopsy: The Last Hours of Natalie Cole." Autopsy. Nar. Eric Meyers. Exec. Prod. Ed Taylor and Michael Kelpie. Reelz, 27 May 2017. Television.
Further reading[]
- Rubin LJ, Badesch DB (August 2005). "Evaluation and management of the patient with pulmonary arterial hypertension". Annals of Internal Medicine. 143 (4): 282–92. CiteSeerX 10.1.1.463.8466. doi:10.7326/0003-4819-143-4-200508160-00009. PMID 16103472. S2CID 28841269.
- Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. (November 2015). "Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society". Circulation. 132 (21): 2037–99. doi:10.1161/CIR.0000000000000329. PMID 26534956. S2CID 7412370.
External links[]
| Classification | |
|---|---|
| External resources |
| Wikimedia Commons has media related to Pulmonary hypertension. |
| Scholia has a topic profile for Pulmonary hypertension. |
| Authority control: National libraries |
|---|
- Pulmonary heart disease and diseases of pulmonary circulation
- Hypertension