Triphenylphosphine

| |

| |

| |

| |
| Names | |
|---|---|
| Preferred IUPAC name
Triphenylphosphane[1] | |
| Identifiers | |
3D model (JSmol)
|
|
| ChEBI | |
| ChemSpider | |
| ECHA InfoCard | 100.009.124 |
| EC Number |
|
PubChem CID
|
|
| RTECS number |
|
| UNII | |
| UN number | 3077 |
CompTox Dashboard (EPA)
|
|
| |
| |
| Properties | |
| C18H15P | |
| Molar mass | 262.292 g·mol−1 |
| Appearance | White Solid |
| Density | 1.1 g cm−3, solid |
| Melting point | 80 °C (176 °F; 353 K) |
| Boiling point | 377 °C (711 °F; 650 K) |
| Insoluble | |
| Solubility | organic solvents |
| Acidity (pKa) | 7.64[2] (pKa of conjugate acid in acetonitrile) |
| -166.8·10−6 cm3/mol | |
Refractive index (nD)
|
1.59; εr, etc. |
| Structure | |
| Pyramidal | |
Dipole moment
|
1.4 - 1.44 D [3] |
| Hazards | |
| Safety data sheet | JT Baker |
| GHS labelling: | |
 
| |
Signal word
|
Danger |
| H302, H317, H350, H412 | |
| P201, P202, P261, P264, P270, P272, P273, P280, P281, P301+P312, P302+P352, P308+P313, P321, P330, P333+P313, P363, P405, P501 | |
| NFPA 704 (fire diamond) | 
2
1
2 |
| Flash point | 180 °C (356 °F; 453 K) |
| Related compounds | |
Related tertiary phosphines
|
Trimethylphosphine Phosphine |
Related compounds
|
Triphenylamine Triphenylarsine Triphenylphosphine oxide Triphenylphosphine sulfide Triphenylphosphine dichloride Triphenylphosphine selenide, Pd(PPh3)4 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
Triphenylphosphine (IUPAC name: triphenylphosphane) is a common organophosphorus compound with the formula P(C6H5)3 and often abbreviated to PPh3 or Ph3P. It is widely used in the synthesis of organic and organometallic compounds. PPh3 exists as relatively air stable, colorless crystals at room temperature. It dissolves in non-polar organic solvents such as benzene and diethyl ether.
Preparation and structure[]
Triphenylphosphine can be prepared in the laboratory by treatment of phosphorus trichloride with phenylmagnesium bromide or phenyllithium. The industrial synthesis involves the reaction between phosphorus trichloride, chlorobenzene, and sodium.:[4]
- PCl3 + 3 PhCl + 6 Na → PPh3 + 6 NaCl
Triphenylphosphine crystallizes in triclinic[5] and monoclinic modification[6] In both cases, the molecule adopts a pyramidal structure with propeller-like arrangement of the three phenyl groups.
Principal reactions with chalcogens, halogens, and acids[]
Oxidation[]
Triphenylphosphine undergoes slow oxidation by air to give triphenylphosphine oxide, Ph3PO:
- 2 PPh3 + O2 → 2 OPPh3
This impurity can be removed by recrystallisation of PPh3 from either hot ethanol or hot isopropanol.[7] This method capitalizes on the fact that OPPh3 is more polar and hence more soluble in polar solvents than PPh3.
Triphenylphosphine abstracts sulfur from polysulfide compounds, episulfides, and elemental sulfur. Simple organosulfur compounds such as thiols and thioethers are unreactive, however. The phosphorus-containing product is triphenylphosphine sulfide, Ph3PS. This reaction can be employed to assay the "labile" S0 content of a sample, say vulcanized rubber. Triphenylphosphine selenide, Ph3PSe, may be easily prepared via treatment of PPh3 with red (alpha-monoclinic) Se. Salts of selenocyanate, SeCN−, are used as the Se0 source. PPh3 can also form an adduct with Te, although this adduct primarily exists as (Ph3P)2Te rather than PPh3Te.[8]
Aryl azides react with PPh3 to give phosphanimines, analogues of OPPh3, via the Staudinger reaction. Illustrative is the preparation of triphenylphosphine phenylimide:
- PPh3 + PhN3 → PhNPPh3 + N2
The phosphanimine can be hydrolyzed to the amine. Typically the intermediate phosphanimine is not isolated.
- PPh3 + RN3 + H2O → OPPh3 + N2 + RNH2
Chlorination[]
Cl2 adds to PPh3 to give triphenylphosphine dichloride ([PPh3Cl]Cl), which exists as the moisture-sensitive phosphonium halide. This reagent is used to convert alcohols to alkyl chlorides in organic synthesis. Bis(triphenylphosphine)iminium chloride (PPN+Cl−, formula [(C6H5)3P)2N]Cl is prepared from triphenylphosphine dichloride:[9]
- 2 Ph3PCl2 + NH2OH·HCl + Ph3P → {[Ph3P]2N}Cl + 4HCl + Ph3PO
Protonation[]
PPh3 is a weak base. It forms isolable triphenylphosphonium salts with strong acids such as HBr:[10]
- P(C6H5)3 + HBr → [HP(C6H5)3]+Br−
Organic reactions[]
PPh3 is widely used in organic synthesis. The properties that guide its usage are its nucleophilicity and its reducing character.[11] The nucleophilicity of PPh3 is indicated by its reactivity toward electrophilic alkenes, such as Michael-acceptors, and alkyl halides. It is also used in the synthesis of biaryl compounds, such as the Suzuki reaction.
Quaternization[]
PPh3 combines with alkyl halides to give phosphonium salts. This quaternization reaction is particularly fast for benzylic and allylic halides:
- PPh3 + CH3I → [CH3PPh3]+I−
These salts, which can often be isolated as crystalline solids, react with strong bases to form ylides, which are reagents in the Wittig reactions.
Aryl halides will quaternize PPh3 to give tetraphenylphosphonium salts:
- PPh3 + PhBr → [PPh4]Br
The reaction however requires elevated temperatures and metal catalysts.
Mitsunobu reaction[]
In the Mitsunobu reaction, a mixture of triphenylphosphine and diisopropyl azodicarboxylate ("DIAD", or its diethyl analogue, DEAD) converts an alcohol and a carboxylic acid to an ester. The DIAD is reduced as it serves as the hydrogen acceptor, and the PPh3 is oxidized to OPPh3.
Appel reaction[]
In the Appel reaction, a mixture of PPh3 and CX4 (X = Cl, Br) is used to convert alcohols to alkyl halides. Triphenylphosphine oxide (OPPh3) is a byproduct.
- PPh3 + CBr4 + RCH2OH → OPPh3 + RCH2Br + HCBr3
This reaction commences with nucleophilic attack of PPh3 on CBr4, an extension of the quaternization reaction listed above.
Deoxygenation[]
The easy oxygenation of PPh3 is exploited in its use to deoxygenate organic peroxides, which generally occurs with retention of configuration:
- PPh3 + RO2H → OPPh3 + ROH (R = alkyl)
It is also used for the decomposition of organic ozonides to ketones and aldehydes, although dimethyl sulfide is more popular for the reaction as the side product, dimethyl sulfoxide is more readily separated from the reaction mixture than triphenylphosphine oxide. Aromatic N-oxides are reduced to the corresponding amine in high yield at room temperature with irradiation:[12]
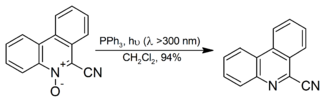
Sulfonation[]
Sulfonation of PPh3 gives tris(3-sulfophenyl)phosphine, P(C6H4-3-SO3−)3 (TPPTS), usually isolated as the trisodium salt. In contrast to PPh3, TPPTS is water-soluble, as are its metal derivatives. Rhodium complexes of TPPTS are used in certain industrial hydroformylation reactions.[13]

Reduction to diphenylphosphide[]
Lithium in THF as well as Na or K react with PPh3 to give Ph2PM (M = Li, Na, K). These salts are versatile precursors to tertiary phosphines.[14][15] For example, 1,2-dibromoethane and Ph2PM react to give Ph2PCH2CH2PPh2. Weak acids such ammonium chloride, convert Ph2PM (M = Li, Na, K) into diphenylphosphine:[15]
- (C6H5)2PM + H2O → (C6H5)2PH + MOH
Transition metal complexes[]
Triphenylphosphine binds well to most transition metals, especially those in the middle and late transition metals of groups 7–10.[16] In terms of steric bulk, PPh3 has a Tolman cone angle of 145°,[17] which is intermediate between those of P(C6H11)3 (170°) and P(CH3)3 (115°). In an early application in homogeneous catalysis, NiBr2(PPh3)2 was used by Walter Reppe for the synthesis of acrylate esters from alkynes, carbon monoxide, and alcohols.[18] The use of PPh3 was popularized by its use in the hydroformylation catalyst RhH(PPh3)3(CO).
Polymer-anchored PPh3 derivatives[]
Polymeric analogues of PPh3 are known whereby polystyrene is modified with PPh2 groups at the para position. Such polymers can be employed in many of the applications used for PPh3 with the advantage that the polymer, being insoluble, can be separated from products by simple filtration of reaction slurries. Such polymers are prepared via treatment of 4-lithiophenyl-substituted polystyrene with chlorodiphenylphosphine (PPh2Cl).
See also[]
References[]
- ^ International Union of Pure and Applied Chemistry (2014). Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013. The Royal Society of Chemistry. p. 431. doi:10.1039/9781849733069. ISBN 978-0-85404-182-4.
- ^ Haav, Kristjan; Saame, Jaan; Kütt, Agnes; Leito, Ivo (2012). "Basicity of Phosphanes and Diphosphanes in Acetonitrile". European Journal of Organic Chemistry. 2012 (11): 2167–2172. doi:10.1002/ejoc.201200009. ISSN 1434-193X.
- ^ Warchol, M.; Dicarlo, E. N.; Maryanoff, C. A.; Mislow, K. (1975). "Evidence for the Contribution of the Lone Pair to the Molecular Dipole Moment of Triarylphosphines". Tetrahedron Letters. 16 (11): 917–920. doi:10.1016/S0040-4039(00)72019-3.
- ^ Corbridge, D. E. C. (1995). Phosphorus: An Outline of its Chemistry, Biochemistry, and Technology (5th ed.). Amsterdam: Elsevier. ISBN 0-444-89307-5.
- ^ Kooijman, H.; Spek, A. L.; van Bommel, K. J. C.; Verboom, W.; Reinhoudt, D. N. (1998). "A Triclinic Modification of Triphenylphosphine" (PDF). Acta Crystallographica. C54 (11): 1695–1698. doi:10.1107/S0108270198009305.
- ^ Dunne, B. J.; Orpen, A. G. (1991). "Triphenylphosphine: a Redetermination" (PDF). Acta Crystallographica. C47 (2): 345–347. doi:10.1107/S010827019000508X.
- ^ Armarego, W. L. F.; Perrin, D. D.; Perrin, D. R. (1980). Purification of Laboratory Chemicals (2nd ed.). New York: Pergamon. p. 455. ISBN 9780080229614.
- ^ Jones, C. H. W.; Sharma, R. D. (1987). "125Te NMR and Mössbauer Spectroscopy of Tellurium-Phosphine Complexes and the Tellurocyanates". Organometallics. 6 (7): 1419–1423. doi:10.1021/om00150a009.
- ^ Ruff, J.K.; Schlientz, W.J. (1974). μ-nitrido-Bis(triphenylphosphorus)(1+ ("PPN") Salts with Metal Carbonyl Anions. Inorg. Synth. Inorganic Syntheses. 15. pp. 84–90. doi:10.1002/9780470132463.ch19. ISBN 9780470132463.
- ^ Hercouet, A.; LeCorre, M. (1988) Triphenylphosphonium bromide: A convenient and quantitative source of gaseous hydrogen bromide. Synthesis, 157–158.
- ^ Cobb, J. E.; Cribbs, C. M.; Henke, B. R.; Uehling, D. E.; Hernan, A. G.; Martin, C.; Rayner, C. M. (2004). "Triphenylphosphine". In L. Paquette (ed.). Encyclopedia of Reagents for Organic Synthesis. New York: J. Wiley & Sons. doi:10.1002/047084289X.rt366.pub2. ISBN 0471936235.
- ^ Burke, S. D.; Danheiser, R. L. (1999). "Triphenylphosphine". Handbook of Reagents for Organic Synthesis, Oxidizing and Reducing Agents. Wiley. p. 495. ISBN 978-0-471-97926-5.
- ^ Herrmann, W. A.; Kohlpaintner, C. W. (1998). Syntheses of Water-Soluble Phosphines and Their Transition Metal Complexes. Inorg. Synth. Inorganic Syntheses. 32. pp. 8–25. doi:10.1002/9780470132630.ch2. ISBN 9780470132630.
- ^ George W. Luther III, Gordon Beyerle (1977). "Lithium Diphenylphosphide and Diphenyl(Trimethylsilyl)Phosphine". Inorganic Syntheses. Inorganic Syntheses. 17. pp. 186–188. doi:10.1002/9780470132487.ch51. ISBN 9780470132487.CS1 maint: uses authors parameter (link)
- ^ a b V. D. Bianco S. Doronzo (1976). "Diphenylphosphine". Inorganic Syntheses. Inorganic Syntheses. 16. pp. 161–188. doi:10.1002/9780470132470.ch43. ISBN 9780470132470.CS1 maint: uses authors parameter (link)
- ^ Elschenbroich, C.; Salzer, A. (1992). Organometallics : A Concise Introduction (2nd ed.). Weinheim: Wiley-VCH. ISBN 3-527-28165-7.
- ^ Immirzi, A.; Musco, A. (1977). "A method to measure the size of phosphorus ligands in coordination complexes". Inorganica Chimica Acta. 25: L41–L42. doi:10.1016/S0020-1693(00)95635-4.
- ^ *Reppe, W.; Schweckendiek, W. J. (1948). "Cyclisierende Polymerisation von Acetylen. III Benzol, Benzolderivate und hydroaromatische Verbindungen". Justus Liebigs Annalen der Chemie. 560 (1): 104–116. doi:10.1002/jlac.19485600104.
External links[]
- Tertiary phosphines
- Phenyl compounds