Nutritional neuroscience

Nutritional neuroscience is the scientific discipline that studies the effects various components of the diet such as minerals, vitamins, protein, carbohydrates, fats, dietary supplements, synthetic hormones, and food additives have on neurochemistry, neurobiology, behavior, and cognition.
Recent research on nutritional mechanisms and their effect on the brain show they are involved in almost every facet of neurological functioning including alterations in neurogenesis, neurotrophic factors, neural pathways and neuroplasticity, throughout the life cycle.[2]
Relatively speaking, the brain consumes an immense amount of energy in comparison to the rest of the body. The human brain is approximately 2% of the human body mass and uses 20–25% of the total energy expenditure.[3] Therefore, mechanisms involved in the transfer of energy from foods to neurons are likely to be fundamental to the control of brain function.[4] Insufficient intake of selected vitamins, or certain metabolic disorders, affect cognitive processes by disrupting the nutrient-dependent processes within the body that are associated with the management of energy in neurons, which can subsequently affect neurotransmission, synaptic plasticity, and cell survival.[4]
Minerals[]
Deficiency or excess of essential minerals (e.g. iron, zinc, copper, and magnesium) can disrupt brain development and neurophysiology to affect behavior.[5] Furthermore, minerals have been implicated in the pathophysiology of neurodegenerative diseases including Alzheimer's dementia.[6][7]
Iron[]
Iron is essential for several critical metabolic enzymes and a deficiency of this mineral can disrupt brain development.[8] For, example chronic marginal iron affects dopamine metabolism and myelin fatty acid composition[9] and behavior in mice.[10] In rats a marginal iron deficiency that does not cause anemia disrupted axon growth in the auditory nerve affecting auditory brainstem latency without major changes in myelination.[11] In rhesus macaques, prenatal iron deficiency disrupts emotional behavior[12] and polymorphisms that reduce the expression of monoamine oxidase interact with gestational iron deficiency to exacerbate the response to a stressful situation leading to increased aggressiveness.[13] Inexpensive and effective iron supplementation is an available preventive strategy recommended by the World Health Organization.[14] However, iron supplementation can exacerbate malaria infection. Therefore, individuals receiving iron supplementation in malaria-endemic areas must be carefully monitored.[15]
Zinc[]
Zinc is essential for the structure and function of thousands of proteins critical for the function of every cell.[16] Zinc can also serve as a neurotransmitter in the brain,[17] thus a deficiency of this mineral can clearly disrupt development as well as neurophysiology. For example, zinc deficiency during early development impairs neurogenesis leading to memory impairments.[18][19] However, zinc deficiency later in life can disrupt appetite and cause depression like behavior.[19][20] However, it is important to consider copper intake relative to zinc supplementation because excess zinc can disrupt copper absorption.[21]
Deficiency[]
Conservative estimates suggest that 25% of the world's population is at risk of zinc deficiency.[22]
Hypozincemia is usually a nutritional deficiency, but can also be associated with malabsorption, diarrhea, acrodermatitis enteropathica, chronic liver disease, chronic renal disease, sickle cell disease, diabetes, malignancy, pyroluria, and other chronic illnesses.[23][24] It can also occur after bariatric surgery, heavy metal exposure[25][26] and tartrazine.[citation needed]
Zinc deficiency is typically the result of inadequate dietary intake of zinc, disease states that promote zinc losses, or physiological states that require increased zinc. Populations that consume primarily plant based diets that are low in bioavailable zinc often have zinc deficiencies.[27][28] Diseases or conditions that involve intestinal malabsorption promote zinc losses. Fecal losses of zinc caused by diarrhea are one contributing factor,[29] often common in developing countries. Changes in intestinal tract absorbability and permeability due, in part, to viral, protozoal, and bacteria pathogens may also encourage fecal losses of zinc.[30] Physiological states that require increased zinc include periods of growth in infants and children as well as in mothers during pregnancy.[31]
Anorexia[]
Zinc deficiency may cause a decrease in appetite which can degenerate into anorexia or anorexia nervosa.[32] Appetite disorders, in turn, cause malnutrition and, notably, inadequate zinc intake. Anorexia itself is a cause of zinc deficiency, thus leading to a vicious cycle: the worsening of anorexia worsens the zinc deficiency. A 1994 randomized, double-blind, placebo-controlled trial showed that zinc (14 mg per day) doubled the rate of body mass increase in the treatment of anorexia nervosa.[33]
Cognitive and motor function impairment[]
Cognitive and motor function may also be impaired in zinc deficient children. Zinc deficiency can interfere with many organ systems especially when it occurs during a time of rapid growth and development when nutritional needs are high, such as during infancy.[34] In animal studies, rats who were deprived of zinc during early fetal development exhibited increased emotionality, poor memory, and abnormal response to stress which interfered with performance in learning situations.[35] Zinc deprivation in monkeys showed that zinc deficient animals were emotionally less mature, and also had cognitive deficits indicated by their difficulty in retaining previously learned problems and in learning new problems.[35] Human observational studies show weaker results. Low maternal zinc status has been associated with less attention during the neonatal period and worse motor functioning.[36] In some studies, supplementation has been associated with motor development in very low birth weight infants and more vigorous and functional activity in infants and toddlers.[36]
Plasma zinc level has been associated with many psychological disorders. However, the nature of this relationship remains unclear in most instances. An increasing amount of evidence suggests that zinc deficiency could play a causal role in the etiology of depression.[37] Indeed, zinc supplementation has been reported to improve measures of depression in randomized double blind placebo controlled trials.[38]
Copper[]
Copper is important for the function of many enzymes in the brain. Notably, dopamine β-mono-oxygenase is affected by copper deficiency leading to increased dopamine and decreased norepinephrine levels.[39] Both copper deficiency and toxicity can interfere with brain development and function.
Deficiency[]
The neurodegenerative syndrome of copper deficiency has been recognized for some time in ruminant animals, in which it is commonly known as "swayback".[40] The disease involves a nutritional deficiency in the trace element copper.[40] Copper is ubiquitous and daily requirement is low making acquired copper deficiency very rare. Copper deficiency can manifest in parallel with vitamin B12 and other nutritional deficiencies.[41] The most common cause of copper deficiency is a remote gastrointestinal surgery, such as gastric bypass surgery, due to malabsorption of copper, or zinc toxicity. On the other hand, Menkes disease is a genetic disorder of copper deficiency involving a wide variety of symptoms that is often fatal.[42]
Neurological presentation[]
Copper deficiency can cause a wide variety of neurological problems including, myelopathy, peripheral neuropathy, and optic neuropathy.[40][43]
Myelopathy[]
Sufferers typically present difficulty walking (gait difficulty) caused by sensory ataxia (irregular muscle coordination) due to dorsal column dysfunction[43] or degeneration of the spinal cord (myelopathy).[40][44] Patients with ataxic gait have problems balancing and display an unstable wide walk. They often feel tremors in their torso, causing side way jerks and lunges.[45]
In brain MRI, there is often an increased T2 signalling at the posterior columns of the spinal cord in patients with myelopathy caused by copper deficiency.[40][43][46] T2 signalling is often an indicator of some kind of neurodegeneration. There are some changes in the spinal cord MRI involving the thoracic cord, the cervical cord or sometimes both.[40][43] Copper deficiency myelopathy is often compared to subacute combined degeneration (SCD).[44] Subacute combined degeneration is also a degeneration of the spinal cord, but instead vitamin B12 deficiency is the cause of the spinal degeneration.[40] SCD also has the same high T2 signalling intensities in the posterior column as copper deficient patient in MRI imaging.[46]
Peripheral neuropathy[]
Another common symptom of copper deficiency is peripheral neuropathy, which is numbness or tingling that can start in the extremities and can sometimes progress radially inward towards the torso.[43][47] In an Advances in Clinical Neuroscience & Rehabilitation (ACNR) published case report, a 69-year-old patient had progressively worsened neurological symptoms.[48] These symptoms included diminished upper limb reflexes with abnormal lower limb reflexes, sensation to light touch and pin prick was diminished above the waist, vibration sensation was lost in the sternum, and markedly reduced proprioception or sensation about the self's orientation.[48] Many people suffering from the neurological effects of copper deficiency complain about very similar or identical symptoms as the patient.[40][47] This numbness and tingling poses danger for the elderly because it increases their risk of falling and injuring themselves. Peripheral neuropathy can become very disabling leaving some patients dependent on wheel chairs or walking canes for mobility if there is lack of correct diagnosis. Rarely can copper deficiency cause major disabling symptoms. The deficiency will have to be present for an extensive amount of time until such disabling conditions manifest.
Optic neuropathy[]
Some patients suffering from copper deficiency have shown signs of vision and color loss.[47] The vision is usually lost in the peripheral views of the eye.[47] The bilateral vision loss is usually very gradual.[47][49] An optical coherence tomography (OCT) shows some nerve fiber layer loss in most patients, suggesting the vision loss and color vision loss was secondary to optic neuropathy or neurodegeneration.[47]
Toxicity[]
Copper toxicity can occur from excessive supplement use, eating acid foods cooked in uncoated copper cookware, exposure to excess copper in drinking water, or as the result of an inherited metabolic disorder in the case of Wilson's disease. A significant portion of the toxicity of copper comes from its ability to accept and donate single electrons as it changes oxidation state. This catalyzes the production of very reactive radical ions, such as hydroxyl radical in a manner similar to Fenton chemistry.[50] This catalytic activity of copper is used by the enzymes with which it is associated, thus is only toxic when unsequestered and unmediated. This increase in unmediated reactive radicals is generally termed oxidative stress, and is an active area of research in a variety of diseases where copper may play an important but more subtle role than in acute toxicity.
Some of the effects of aging may be associated with excess copper.[51] In addition, studies have found that people with mental illnesses, such as schizophrenia, had heightened levels of copper in their systems. However, it is unknown at this stage whether the copper contributes to the mental illness, whether the body attempts to store more copper in response to the illness, or whether the high levels of copper are the result of the mental illness.[52]
Alzheimer's disease[]
Elevated free copper levels exist in Alzheimer's disease.[53] Copper and zinc are known to bind to amyloid beta proteins in Alzheimer's disease.[54] This bound form is thought to mediate the production of reactive oxygen species in the brain.[55] A preliminary clinical trial suggests that zinc supplementation may be able to decrease copper levels and slow degeneration in Alzheimer's disease.[56]
Manganese[]
Manganese is a component of some enzymes and stimulates the development and activity of other enzymes. Manganese superoxide dismutase (MnSOD) is the principal antioxidant in mitochondria. Several enzymes activated by manganese contribute to the metabolism of carbohydrates, amino acids, and cholesterol.[57]
Deficiency of manganese causes skeletal deformation in animals and inhibits the production of collagen in wound healing.[58] On the other hand, manganese toxicity is associated with neurological complications.[59]
Toxicity[]
Manganese poisoning is a toxic condition resulting from chronic exposure to manganese and first identified in 1837 by .[60]
Presentation[]
Chronic exposure to excessive Mn levels can lead to a variety of psychiatric and motor disturbances, termed manganism. Generally, exposure to ambient Mn air concentrations in excess of 5 mg Mn/m3 can lead to Mn-induced symptoms.[61]
In initial stages of manganism, neurological symptoms consist of reduced response speed, irritability, mood changes, and compulsive behaviors.[59] Upon protracted exposure symptoms are more prominent and resemble those of idiopathic Parkinson's disease, as which it is often misdiagnosed, although there are particular differences in both the symptoms (nature of tremors, for example), response to drugs such as levodopa, and affected portion of the basal ganglia. Symptoms are also similar to Lou Gehrig's disease and multiple sclerosis.
Causes[]
Manganism has become an active issue in workplace safety as it has been the subject of numerous product liability lawsuits against manufacturers of arc welding supplies. In these lawsuits, welders have accused the manufacturers of failing to provide adequate warning that their products could cause welding fumes to contain dangerously high manganese concentrations that could lead welders to develop manganism. Companies employing welders are also being sued, for what colloquially is known as "welders' disease". However, studies fail to show any link between employment as a welder and manganism (or other neurological problems).[62][63][64]
Manganism is also documented in reports of illicit methcathinone manufacturing.[65] This is due to manganese being a byproduct of methcathinone synthesis if potassium permanganate is used as an oxidiser.[66] Symptoms include apathy, bradykinesia, gait disorder with postural instability, and spastic-hypokinetic dysarthria. Another street drug sometimes contaminated with manganese is the so-called "Bazooka", prepared by free-base methods from cocaine using manganese carbonate.[67]
Reports also mention such sources as contaminated drinking water,[68] and fuel additive methylcyclopentadienyl manganese tricarbonyl (MMT),[69] which on combustion becomes partially converted into manganese phosphates and sulfate that go airborne with the exhaust,[70][71][72] and manganese ethylene-bis-dithiocarbamate (Maneb), a pesticide.[73]
Pathological mechanisms[]
Manganese may affect liver function, but the threshold of acute toxicity is very high. On the other hand, more than 95% of manganese is eliminated by biliary excretion. Any existing liver damage may slow this process, increasing its concentration in blood plasma.[74] The exact neurotoxic mechanism of manganese is uncertain but there are clues pointing at the interaction of manganese with iron,[75][76][77][78] zinc,[79] aluminum,[75][79] and copper.[79] Based on a number of studies, disturbed iron metabolism could underlie the neurotoxic action of manganese.[80]
It participates in Fenton reactions and could thus induce oxidative damage, a hypothesis corroborated by the evidence from studies of affected welders.[81] A study of the exposed workers showed that they have significantly fewer children.[82] This may indicate that long-term accumulation of manganese affects fertility. Pregnant animals repeatedly receiving high doses of manganese bore malformed offspring significantly more often compared to controls.[83] Manganism mimics Schizophrenia.[84] It is found in large quantities in paint and steelmaking.
Treatment[]
The current mainstay of manganism treatment is levodopa and chelation with EDTA. Both have limited and at best transient efficacy. Replenishing the deficit of dopamine with levodopa has been shown to initially improve extrapyramidal symptoms,[85][86][87] but the response to treatment goes down after 2 or 3 years,[88] with worsening condition of the same patients noted even after 10 years since last exposure to manganese.[89] Enhanced excretion of manganese prompted by chelation therapy brings its blood levels down but the symptoms remain largely unchanged, raising questions about efficacy of this form of treatment.[90][91]
Increased ferroportin protein expression in human embryonic kidney (HEK293) cells is associated with decreased intracellular Mn concentration and attenuated cytotoxicity, characterized by the reversal of Mn-reduced glutamate uptake and diminished lactate dehydrogenase (LDH) leakage.[61]
Locations[]
The Red River Delta near Hanoi has high levels of manganese or arsenic in the water. Approximately 65 percent of the region's wells contain high levels of arsenic, manganese, selenium and barium.[92] This was also published in the Proceedings of the National Academy of Sciences.
Magnesium[]
Magnesium is necessary for the function of many metabolic enzymes and also serves as a key regulator of calcium channels involved in neurotransmission (e.g. NMDA receptor).[93] Magnesium supplementation facilitates nerve regeneration after injury.[94] Although unpolished grains contain magnesium, phytic acid in grains can inhibit its absorption. Leafy greens are an excellent source of magnesium.[95]
Vitamins[]
Deficiency or excess intake of many vitamins can affect the brain contributing to developmental[96] and degenerative diseases.[97]
Vitamin A[]
Vitamin A is an essential nutrient for mammals which takes form in either retinol or the provitamin beta-Carotene. It helps regulation of cell division, cell function, genetic regulation, helps enhance the immune system, and is required for brain function, chemical balance, growth and development of the central nervous system and vision.[citation needed]
Learning memory[]
In an experiment by Chongqing Medical University pregnant rats were either plentiful in vitamin A or were of a vitamin A deficiency (VAD) due to their diet. The offspring of these rats were then tested in a water maze at 8 weeks old and it was found the VAD offspring had a harder time finishing the maze which helps show that these rats, even while having a deficiency from in utero, have more problems with learning memory.[98] Young rats in a separate study by the same university also showed impaired long-term potentiation in the hippocampus when they were VAD which shows neuronal impairment.[99] When the patient is VAD for too long, the effects of the damage to the hippocampus can be irreversible.[100]
Spatial memory[]
Vitamin A affects spatial memory most of the time because the size of the nuclei in hippocampal neurons are reduced by approximately 70% when there is a deficiency which affects a person's abilities for higher cognitive function. In a study by the University of Cagliari, Italy, VAD rats had more trouble learning a Radial arm maze than rats who had normal levels of the vitamin. The healthy rats were able to correctly solve the maze within the 15-day training period and other rats that were once deficient but had vitamin A restored to normal levels were also able to solve it. Here it was found that the retinoid receptors which help transport vitamin A were of normal function.[101]
Prevention, treatment and symptoms[]
Eating foods high in vitamin A or taking dietary supplements, retinol or retinal will prevent a deficiency. The foods highest in vitamin A are any pigmented fruits and vegetables and leafy green vegetables also provide beta-Carotene.[citation needed] There can be symptoms of fat loss and a reduction of any weight gain that would be considered normal for an individual,[101] especially developmental weight gains such as in infants which would occur if the infant was deprived of vitamin A in utero and/or if it was deprived postnatal for an extensive period of time.[98] The deficiency can also cause conditions such as blindness or night blindness, also known as nyctalopia. Night blindness is due to the inability to regenerate rhodopsin in the rods which is needed in dim light in order to see properly.[citation needed] A treatment of supplements of retinoic acid which is a part of vitamin A can help replenish levels and help bring learning to normal,[102] but after 39 weeks this is ineffective even if the treatment is daily because it will not bring the retinoid hypo-signalling back to normal.[100]
Relationship with zinc[]
Zinc is needed to maintain normal vitamin A levels in blood plasma.[citation needed] It also helps vitamin A become metabolized by the liver. However evidence suggests that when someone is deficient in both vitamin A and zinc, memory is more improved when just vitamin A is increased than when just zinc is increased. Of course memory has the largest improvement when both are increased. When one of these nutrients is not balanced, the other is most likely to be affected because they rely on each other for proper functioning in learning.[103]
Thiamin (vitamin B1)[]
Vitamin B1, also known as thiamine, is a coenzyme essential for the metabolism of carbohydrates.[104] This vitamin is important for the facilitation of glucose use, thus ensuring the production of energy for the brain,[105] and normal functioning of the nervous system, muscles, and heart.[106]
Thiamine is found in all living tissues,[107] and is uniformly distributed throughout mammalian nervous tissue, including the brain and spinal cord. Metabolism and coenzyme function of the vitamin suggest a distinctive function for thiamin within the nervous system.[108]
The brain retains its thiamine content in the face of a vitamin-deficient diet with great tenacity, as it is the last of all nervous tissues studied to become depleted. A 50% reduction of thiamine stores in rats becomes apparent after only 4 days of being put on a thiamine-deficient diet. However, polyneuritic signs do not begin to appear until about 4 or 5 weeks have passed.[108] Similar results have been found in human subjects.[107]
Deficiencies[]
The body has only small stores of B1; accordingly, there is risk of deficiency if the level of intake is reduced only for a few weeks.[107] Thiamin deficiency during critical periods of early development can disrupts neurogenesis in animal models.[109] Lack of thiamin later in life causes the disease known as beriberi.[110] There are two forms of beriberi: "wet", and "dry". Dry beriberi is also known as cerebral beriberi. Characteristics of wet beriberi include prominent edema and cardiac involvement, whereas dry beriberi is mainly characterized by a polyneuritis.[108] Severe thiamin deficiency can also result in acute neurodegeneration leading to peripheral neuropathy and memory loss.[111]
In industrialized nations, thiamine deficiency is a clinically significant problem in individuals with chronic alcoholism or other disorders that interfere with normal ingestion of food.[112] Thiamine deficiency within developed nations tends to manifest as Wernicke–Korsakoff syndrome.[110] Chronic alcoholism can disrupt thiamin absorption and thiamin deficiency contributes to neurodegeneration and memory loss in alcoholics known as Wernicke's encephalopathy.[113] Individuals with chronic alcoholism may fall short on minimum daily requirements of thiamine in part due to anorexia, erratic eating habits, lack of available food, or a combination of any of these factors. Thiamine deficiency has been reported in up to 80% of alcoholic patients due to inadequate nutritional intake, reduced absorption, and impaired utilization of thiamine.[114] Alcohol, in combination with its metabolite acetaldehyde, interacts with thiamine utilization at the molecular level during transport, diphosphorylation, and modification processes. For this reason, chronic alcoholics may have insufficient thiamine for maintenance of normal brain function, even with seemingly adequate dietary intake.[112]
Symptoms[]
Clinical signs of B1 deficiency include mental changes such as apathy, decrease in short-term memory, confusion, and irritability.[110] Moderate deficiency in thiamine may reduce growth in young populations, in increase chronic illness in both young and middle-aged adults. In addition, moderate deficiency of thiamine may increase rates of depression, dementia, falls, and fractures in old age.[112]
The lingering symptoms of neuropathy associated with cerebral beriberi are known as Korsakoff's syndrome, or the chronic phase of Wernicke-Korsakoff's.[115] Wernicke encephalopathy is a neurological disorder resulting from a deficiency in thiamine, sharing the same predominant features of cerebral beriberi, as characterized by ocular abnormalities, ataxia of gait, a global state of confusion, and neuropathy.[112] The state of confusion associated with Wernicke's may consist of apathy, inattention, spatial disorientation, inability to concentrate, and mental sluggishness or restlessness.[104] Clinical diagnosis of Wernicke's disease cannot be made without evidence of ocular disturbance, yet these criteria may be too rigid.[116] Korsakoff's likely represents a variation in the clinical manifestation of Wernicke encephalophathy, as they both share similar pathological origin.[116]
Korsakoff's syndrome is often characterized by confabulation, disorientation, and profound amnesia.[115] Characteristics of the neuropathology are varied, but generally consist of bilaterally symmetrical midline lesions of brainstem areas, including the mammillary bodies, thalamus, periaqueductal region, hypothalamus, and the cerebellar vermis.[112][115]
Treatment[]
Immediate treatment of Wernicke encephalopathy involves the administration of intravenous thiamine, followed with long-term treatment and prevention of the disorder through oral thiamine supplements, alcohol abstinence, and a balanced diet.[104] Improvements in brain functioning of chronic alcoholics may occur with abstinence-related treatment, involving the discontinuation of alcohol consumption and improved nutrition.[112] Wernicke's encephalopathy is life-threatening if left untreated. However, a rapid reversal of symptoms may result from prompt administration of thiamine.[107]
Prevention[]
Fortification of flour is practiced in some countries to replace the thiamine lost during processing. However, this method has been criticized for missing the target population of chronic alcoholics, who are most at risk for deficiency. Alternative solutions have suggested the fortification of alcoholic beverages with thiamine.[107]
Ingesting a diet rich in thiamine may stave off the adverse effects of deficiency. Foods providing rich sources of thiamine include unrefined grain products, ready-to-eat cereals, meat (especially pork), dairy products, peanuts, legumes, fruits and eggs.[117]
Niacin (vitamin B3)[]
Vitamin B3, also known as niacin, includes both nicotinamide as well as nicotinic acid, both of which function in many biological oxidization and reduction reactions within the body. These functions include the biochemical degradation of carbohydrates, fats and proteins. Niacin is also involved in the synthesis of fatty acids and cholesterol,[118] which are known mediators of brain biochemistry, and in effect, of cognitive function.[119]
Sufficient niacin intake is either obtained from diet, or synthesized from the amino acid tryptophan.[118]
Deficiencies[]
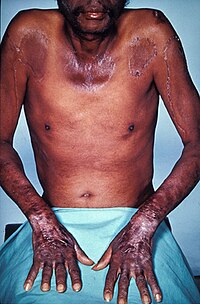
Severe niacin deficiency typically manifests itself as the disease pellagra.[118] Synthesis of B3 from tryptophan involves vitamin B2 and B6, so deficiencies in either of these nutrients can lead to niacin deficiency. An excess of leucine, an essential amino acid, in the diet can also interfere with tryptophan conversion and subsequently result in a B3 deficiency.[120]
Pellagra is most common to populations within developing countries in which corn is the dietary staple. The disease has virtually disappeared from industrialized countries, yet still appears in India and parts of China and Africa.[118] This is in part due to the bound form of niacin that unprocessed corn contains, which is not readily absorbed into the human body. The processes involved in making corn tortillas, can release the bound niacin into a more absorbable form. Pellagra is not problematic in countries which traditionally prepare their corn in this way, but is a problem in other countries where unprocessed corn is main source of caloric intake.[121]
Though pellagra predominantly occurs in developing countries, sporadic cases of pellagra may be observed within industrialized nations, primarily in chronic alcoholics and patients living with functional absorption complications.[120]
Symptoms[]
Pellagra is classically characterized by four 4 "D's": diarrhea, dermatitis, dementia, and death.[120] Neuropsychiatric manifestations of pellagra include headache, irritability, poor concentration, anxiety, hallucinations, stupor, apathy, psychomotor unrest, photophobia, tremor, ataxia, spastic paresis, fatigue, and depression. Symptoms of fatigue and insomnia may progress to encephalopathy characterized by confusion, memory loss, and psychosis.[120]
Those afflicted with pellagra may undergo pathological alterations in the nervous system. Findings may include demylenation and degeneration of various affected parts of the brain, spinal cord, and peripheral nerves.[122]
Treatment[]
Prognosis of deficiency is excellent with treatment. Without, pellagra will gradually progress and lead to death within 4–5 years, often a result of malnutrition from prolonged diarrhea, or complications as caused by concurrent infections or neurological symptoms. Symptoms of pellagra can be cured with exogenous administration of nicotinic acid or nicotinamide.[120]
Flushing occurs in many patients treated therapeutically with nicotinic acid,[123] and as a result, nicotinamide holds more clinical value as it is not associated with the same uncomfortable flushing. The adult dose of nicotinamide is 100 mg taken orally every 6 hours until resolution of major acute symptoms, followed with oral administration of 50 mg every 8–12 hours until skin lesions heal. For children, treatment involves oral ingestion of 10–15 mg of nicotinamide, depending on weight, every 6 hours until signs and symptoms are resolved. Severe cases require 1 gram every 3–4 hours, administered parenterally.[120]
Oral nicotinamide has been promoted as an over-the-counter drug for the treatment of Alzheimer's dementia. Conversely, no clinically significant effect has been found for the drug, as nicotinamide administration has not been found to promote memory functions in patients with mild to moderate dementia of either Alzheimer's, vascular, or fronto-temporal types. This evidence suggests that nicotinamide may treat dementia as related to pellagra, but administration does not effectively treat other types of dementia.[124]
Prevention[]
The best method of prevention is to eat foods rich in B3. Generally, this involves the intake of a protein-rich diet. Foods that contain high concentrations of niacin in the free form include beans and organ meat, as well as enriched grain and cereal products.[118] While niacin is present in corn and other grains, the bioavailability of the nutrient is much less than it is in protein-rich sources. Different methods of processing corn may result in a higher degree of bioavailability of the vitamin.[121]
Though treatment with niacin does little to alter the effects of Alzheimer's dementia, niacin intake from foods is inversely associated with the disease.[125]
Folate (vitamin B9)[]
Folate deficiency can disrupt neurulation and neurogenesis. Maternal folic acid intake around the time of conception prevents neural tube defects.[126] Furthermore, folic acid intake was recently associated with incidence of autism.[127] Enriched white flour is fortified with folic acid in the United States and many other countries. However the European Union does not have mandatory folic acid fortification. Although the protective effects of folic acid are well documented, there remains legitimate concern that fortification could lead to toxic levels in a subset of the population. For example, elevated levels of folic acid can interact with vitamin B12 deficiency to cause neurodegeneration.[128] Furthermore, folic acid and iron can interact to exacerbate malaria.[129]
Folic acid is the most oxidized and stable form of folate, and can also be referred to as vitamin B9. It rarely occurs naturally in foods, but it is the form used in vitamin supplements as well as fortified food products.[130]
Folate coenzymes are involved in numerous conversion processes within the body, including DNA synthesis and amino acid interconversions.[130] Folate and vitamin B12 play a vital role in the synthesis of S-adenosylmethionine, which is of key importance in the maintenance and repairment of all cells, including neurons.[131] In addition, folate has been linked to the maintenance of adequate brain levels of cofactors necessary for chemicals reactions that lead to the synthesis of serotonin and catecholamine neurotransmitters.[130]
Folate has a major, but indirect role in activities which help to direct gene expression and cell proliferation. These activities occur at a greatly increased rate during pregnancy, and depend on adequate levels of folate within blood plasma.[132]
Concentrations of blood plasma folate and homocysteine concentrations are inversely related, such that an increase in dietary folate decreases homocysteine concentration. Thus, dietary intake of folate is a major determinant of homocysteine levels within the body.[133]
Autoantibodies against folate receptor alpha have been found in up to 75% of children with autism.[134]
Deficiencies[]
Folate deficiency most commonly arises from insufficient folate intake from the diet, but may also stem from inefficient absorption or metabolic utilization of folate, usually a result of genetic variation.[135] The relationship between folate and B12 is so interdependent that deficiency in either vitamin can result in megaloblastic anemia, characterized by organic mental change.[136]
The process of neural tube transformation into structures that will eventually develop into the central nervous system is known as neurulation, the success of which is dependent on the presence of folate within the body. This process begins in the human approximately 21 days after conception, and is completed by 28 days. Thus, a woman may not even be aware of her pregnancy by the time the process of neurulation is complete, potentially causing severe consequences in the development of the fetus.[130]
Functional problems in the absorption and utilization of vitamins may also play a role in folate deficiencies within the elderly.[131]
Symptoms[]
The link between levels of folate and altered mental function is not large, but is sufficient enough to suggest a causal association.[130] Deficiency in folate can cause an elevation of homocysteine within the blood,[133] as the clearance of homocysteine requires enzymatic action dependent on folate, and to a lesser extent, vitamins B6 and B12. Elevated homocysteine has been associated with increased risk of vascular events, as well as dementia.[138]
Differences lie in the presentation of megaloblastic anemia induced by either folate or B12 deficiency. Megaloblastic anemia related to deficiency in B12 generally results in peripheral neuropathy, whereas folate-related anemia often results in affective, or mood disorders.[136][139] Neurological effects are not often associated with folate-related megaloblastic anemia, although demyelinating disorders may eventually present.[136] In one study, mood disturbances were recorded for the majority of patients presenting with megaloblastic anemia in the absence of B12 deficiency.[130] In addition, folate concentrations within blood plasma have been found to be lower in patients with both unipolar and bipolar depressive disorders when compared with control groups. In addition, depressive groups with low folate concentrations responded less well to standard antidepressant therapy than did those with normal levels within plasma.[130] However, replication of these findings are less robust.[140]
The role of folic acid during pregnancy is vital to normal development of the nervous system in the fetus. A deficiency in folate levels of a pregnant woman could potentially result in neural tube disorder, a debilitating condition in which the tubes of the central nervous system do not fuse entirely.[132] NTDs are not to be confused with spina bifida, which does not involve neural elements.[130] Neural tube defects can present in a number of ways as a result of the improper closure at various points of the neural tube. The clinical spectrum of the disorder includes encephalocele, craniorachischisis, and anencephaly. In addition, these defects can also be classified as open, if neural tissue is exposed or covered only by membrane, or can be classified as closed, if the tissue is covered by normal skin.[137]
Intake of the vitamin has been linked to deficits in learning and memory, particularly within the elderly population.[130] Elderly people deficient in folate may present with deficits in free recall and recognition, which suggests that levels of folate may be related to efficacy of episodic memory.[141]
Treatment[]
Lack of adequate folate may produce a form of dementia considered to be reversible with administration of the vitamin. Indeed, there is a degree of improvement in memory associated with folate treatment. In a 3-year longitudinal study of men and women aged 50–70 years with elevated homocysteine plasma concentration, researchers found that a daily oral folic acid supplementation of 800μg resulted in an increase in folate levels and a decrease in homocysteine levels within blood plasma. In addition to these results, improvements of memory, and information-processing speed, as well as slight improvements of sensorimotor speed were observed,[142] which suggests there is a link between homocysteine and cognitive performance.
However, while the amount of cognitive improvement after treatment with folate is correlated with the severity of folate deficiency, the severity of cognitive decline is independent of the severity of folate deficiency. This suggests that the dementia observed may not be entirely related to levels of folate, as there could be additional factors that were not accounted for which might have an effect.[143]
Prevention[]
Because neurulation may be completed before pregnancy is recognized, it is recommended that women capable of becoming pregnant take about 400μg of folic acid from fortified foods, supplements, or a combination of the two in order to reduce the risk of neural tube defects.[130] These major anomalies in the nervous system can be reduced by 85% with systematic folate supplementation occurring before the onset of pregnancy.[132]
The incidence of Alzheimer's and other cognitive diseases has been loosely connected to deficiencies in folate. It is recommended for the elderly to consume folate through food, fortified or not, and supplements in order to reduce risk of developing the disease.[143] Good sources of folate include liver, ready-to-eat breakfast cereals, beans, asparagus, spinach, broccoli, and orange juice.[144]
Choline[]
Choline is an important methyl donor involved in one-carbon metabolism that also becomes incorporated into phospholipids and the neurotransmitter acetylcholine. Because of its role in cellular synthesis, choline is an important nutrient during the prenatal and early postnatal development of offspring as it contributes heavily to the development of the brain. A study found that rats that were given supplements of choline in utero or in the weeks following birth had superior memories. The changes appeared to be a result of physical changes to the hippocampus, the area of the brain responsible for memory.[145][146] Furthermore, choline can reduce some of the deleterious effects of folate deficiency on neurogenesis.[147]
While choline during development is important, adult levels of choline are also important. Choline has been shown to increase the synthesis and release of acetylcholine from neurons,[148] which in turn increases memory. A double-blind study was conducted using normal college students (no neurological disorders). Results showed that twenty-five grams of phosphatidylcholine (another form of choline) created a significant improvement in explicit memory, measured by a serial learning task, however this improvement may be attributed to the improvement of slow learners.[149] Another study found that a single ten-gram oral dose of choline, given to normal volunteers (again, without neurological disorders) significantly decreased the number of trials needed to master a serial-learning word test. This increase in memory is particularly beneficial to memory loss suffered by old age. A study conducted on rats who, like humans, suffer from an age-related loss of memory were tested on how choline affected memory. The results showed that rats who had a chronic low-choline diet showed greater memory loss then their same-age control counterparts, while rats who had choline-enriched diets showed a diminished memory loss compared to both the choline-low diet and control rat groups. Furthermore, young rats who were choline-deficient performed as poorly on memory tasks as older rats while older rats that were given choline supplements performed as well as three-month-old rats.[150]
Deficiencies and treatments[]
Despite the wide range of foods that choline is found in, studies have shown that the mean choline intake of men, women and children are below the Adequate Intake levels.[151] It is important to note that not enough choline is naturally produced by the body, so diet is an important factor. People who consume a large quantity of alcohol may be at an increased risk for choline deficiency. Sex and age also plays a role, with premenopausal females being less sensitive to choline deficiency than either males or postmenopausal females.[150] This has been theorized to be a result of premenopausal women having an increased ability to synthesize choline in some form, which has been confirmed in studies on rats.[152] In such instances of deficiency, choline supplements or (if able) dietary changes may be beneficial. Good sources of choline include liver, milk, eggs and peanuts.[153] There is further evidence to suggest that choline supplements can be used to treat people who suffer from neurological disorders as well we memory defects.[150][151] Oral doses of CDP-choline (another form of choline) given to elderly subjects with memory deficits, but without dementia, for four weeks showed improved memory in free recall tasks, but not in recognition tests.[154] In a second study, patients with early Alzheimer-type dementia were treated with twenty-give gram doses of phosphatidylcholine every day for six months. Slightly improvements were shown in memory tests compared to the placebo control group. Other studies conducted did not find any such improvement.
Cobalamin (vitamin B12)[]
Also known as cobalamin, B12 is an essential vitamin necessary for normal blood formation. It is also important for the maintenance of neurological function and psychiatric health.[155] The absorption of B12 into the body requires adequate amounts of intrinsic factor, the glycoprotein produced in the parietal cells of the stomach lining. A functioning small intestine is also necessary for the proper metabolism of the vitamin, as absorption occurs within the ileum.[155]
B12 is produced in the digestive tracts of all animals, including humans.[156] Thus, animal-origin food is the only natural food source of vitamin B12[157] However, synthesis of B12 occurs in the large intestine, which is past the point of absorption that occurs within the small intestine. As such, vitamin B12 must be obtained through diet.[156]
Deficiencies[]
Unlike other B vitamins which are not stored in the body, B12 is stored in the liver. Because of this, it may take 5–10 years before a sudden dietary B12 deficiency will become apparent in a previously healthy adult.[158] B12 deficiency, also known as hypocobalaminemia, often results from complications involving absorption into the body.[159]
B12 deficiency is often associated with pernicious anemia, as it is the most common cause. Pernicious anemia results from an autoimmune disorder which destroys the cells that produce intrinsic factor within the stomach lining, thereby hindering B12 absorption. B12 absorption is important for the subsequent absorption of iron, thus, people with pernicious anemia often present with typical symptoms of anemia, such as pale skin, dizziness, and fatigue.[160]
Among those at highest risk for B12 deficiency are the elderly population, as 10-15% of people aged 60+ may present with some form of hypocobalaminemia. High rates of deficiency in the elderly commonly results from the decrease of functional absorption of B12, as production of intrinsic factor declines with age. However, pernicious anemia is the most common cause of B12 deficiency in North American and European populations.[157]
Those afflicted with various gastrointestinal diseases may also be at risk for deficiency as a result of malabsorption. These diseases may affect production of intrinsic factor in the stomach, or of pancreatic bile. Diseases that involve disorders of the small intestine, such as celiac disease, Crohn's disease and ileitis, may also reduce B12 absorption. For example, people with celiac disease may damage the microvilli within their small intestines through the consumption of gluten, thereby inhibiting absorption of B12 as well as other nutrients.[159]
A diet low in B12, whether voluntary or not, can also cause symptoms of hypocobalaminemia. Many rich sources of B12 come from animal meats and by-products. Populations in developing countries may not have access to these foods on a consistent basis, and as a result may become deficient in B12.[161] In addition, vegans, and to a lesser extent vegetarians, are at risk for consuming a diet low in cobalamin as they voluntarily abstain from animal sources of B12.[159] A combination of these two scenarios may increase prevalence of cobalamin deficit. For instance, B12 deficiency is problematic in India, where the majority of the population is vegetarian and the scarcity of meat consumption is common for omnivores as well.[161]
Symptoms[]
An assortment of neurological effects can be observed in 75-90% of individuals of any age with clinically observable B12 deficiency. Cobalamin deficiency manifestations are apparent in the abnormalities of the spinal cord, peripheral nerves, optic nerves, and cerebrum. These abnormalities involve a progressive degeneration of myelin,[162] and may be expressed behaviourally through reports of sensory disturbances in the extremities, or motor disturbances, such as gait ataxia. Combined myelopathy and neuropathy are prevalent within a large percentage of cases. Cognitive changes may range from loss of concentration to memory loss, disorientation, and dementia. All of these symptoms may present with or without additional mood changes.[157] Mental symptoms are extremely variable, and include mild disorders of mood, mental slowness, and memory defect. Memory defect encompasses symptoms of confusion, severe agitation and depression, delusions and paranoid behaviour, visual and auditory hallucinations, urinary and fecal incontinence in the absence of overt spinal lesions, dysphasia, violent maniacal behaviour, and epilepsy. It has been suggested that mental symptoms could be related to a decrease in cerebral metabolism, as caused by the state of deficiency.[162] All of these symptoms may present with or without additional mood changes.[157]
Mild to moderate cases of pernicious anemia may show symptoms of bleeding gums, headache, poor concentration, shortness of breath, and weakness. In severe cases of pernicious anemia, individuals may present with various cognitive problems such as dementia, and memory loss.[160]
It is not always easy to determine whether B12 deficiency is present, especially within older adults.[159] Patients may present with violent behaviour or more subtle personality changes. They may also present with vague complaints, such as fatigue or memory loss, that may be attributed to normative aging processes. Cognitive symptoms may mimic behaviour in Alzheimer's and other dementias as well.[157] Tests must be run on individuals presenting with such signs to confirm or negate cobalamin deficiency within the blood.[160]
Treatment[]

Patients deficient in B12 despite normal absorption functionality may be treated through oral administration of at least 6 mg of the vitamin in pill form. Patients who suffer from irreversible causes of deficiency, such as pernicious anemia or old age, will need lifelong treatment with pharmacological doses of B12. Strategy for treatment is dependent on the patient's level of deficiency as well as their level of cognitive functioning.[159] Treatment for those with severe deficiency involves 1000 mg of B12 administered intramuscularly daily for one week, weekly for one month, then monthly for the rest of the patient's life. Daily oral supplementation of B12 mega-doses may be sufficient in reliable patients, but it is imperative that the supplementation be continued on a lifelong basis as relapse may occur otherwise.[160]
The progression of neurological manifestations of cobalamin deficiency is generally gradual. As a result, early diagnosis is important or else irreversible damage may occur.[155] Patients who become demented usually show little to no cognitive improvement with the administration of B12.[160]
A deficiency in folate may produce anemia similar to the anemia resulting from B12 deficiency. There is risk that folic acid administered to those with B12 deficiency may mask anemic symptoms without solving the issue at hand. In this case, patients would still be at risk for neurological deficits associated with B12 deficiency-related anemia, which are not associated with anemia related to folate deficiency.[135]
Prevention[]
In addition to meeting intake requirements through food consumption, supplementation of diet with vitamin B12 is seen as a viable preventive measure for deficiency. It has been recommended for the elderly to supplement 50 mcg a day in order to prevent deficit from occurring.[160]
Animal protein products are a good source of B12, particularly organ meats such as kidney or liver. Other good sources are fish, eggs, and dairy products.[156] It is suggested that vegans, who consume no animal meat or by-products, supplement their diet with B12. While there are foods fortified with B12 available, some may be mislabelled in an attempt to boost their nutritional claims. Products of fermentation, such as algae extracts and sea vegetables, may be labelled as sources of B12, but actually contain B12 analogues which compete for the absorption of the nutrient itself.[161] In order to get adequate amounts of the vitamin, orally administered pills or fortified foods such as cereals and soy milk, are recommended for vegans.[163]
Vitamin D[]
Vitamin D is an essential regulator of the vitamin D receptor that controls gene transcription during development. The vitamin D receptor is strongly expressed in the substantia nigra.[164] Accordingly, vitamin D deficiency can disrupt neurogenesis leading to altered dopamine signaling and increased exploratory behavior in rats.[165][166] This is considered a rodent model of the schizophrenia phenotype and vitamin D deficiency has been proposed as an explanation for the increased incidence of schizophrenia among children that were conceived during winter months. A Finnish study found that vitamin D supplement use is associated with reduced risk of schizophrenia.[167]
Lipids[]
Fat[]
Fatty acids are necessary for the synthesis of cell membranes neurotransmitters and other signaling molecules. While excessive fat intake can be harmful, deficiency of essential fatty acids can disrupt neurodevelopment and synaptic plasticity.[168]
Saturated fat[]
Consuming large amounts of saturated fat can negatively affect the brain. Eating foods with saturated fats elevates the level of cholesterol and triglycerides in the body. Studies have shown that high levels of triglycerides strongly link with mood problems such as depression, hostility and aggression. This may occur because high triglyceride levels decrease the amount of oxygen that blood can carry to the brain.[169] The American Heart Association recommends that people consume no more than 16g of saturated fat daily. Common sources of saturated fat are meat and dairy products.
Essential fatty acids[]
There are two kinds of essential fatty acids that people must consume (omega-3 and omega-6). Many academics recommend a balanced amount of omega-3s and omega-6s. However, some estimate that Americans consume twenty times more omega-6s than omega-3s. There is a theory that an imbalance of essential fatty acids may lead to mental disorders such as depression, hyperactivity and schizophrenia, but it still lacking evidences. An omega-3 deficient diet increases omega-6 levels in the brain disrupting endocannabinoid signaling in the prefrontal cortex and nucleus accumbens contributing to anxiety and depression-like behaviors in mice.[168] Sources of omega-3 include flax seeds, chia seeds, walnuts, sea vegetables, green leafy vegetables, and cold water fish. Sources of omega-6 include walnuts, hazelnuts; sunflower, safflower, corn, and sesame oils.[170]
Cholesterol[]
While cholesterol is essential for membranes and steroid hormones, excess cholesterol affects blood flow impairing cognitive function in vascular dementia.[171]
Carbohydrates[]
Studies have shown that learning and memory improve after consuming carbohydrates. There are two kinds of carbohydrates people consume: simple and complex. Simple carbohydrates are often found in processed foods and release sugar into the bloodstream quickly after consumption. Complex carbohydrates are digested more slowly and therefore cause sugar to be released into the bloodstream more slowly.[172] Good sources of complex carbohydrates are whole-grain breads, pasta, brown rice, oatmeal, and potatoes. It is recommended that people consume more complex carbohydrates because consuming complex carbohydrates will cause the level of sugar in the bloodstream to be more stable, which will cause less stress hormones to be released. Consuming simple carbohydrates may cause the levels of sugar in the bloodstream to rise and fall, which can cause mood swings.[173]
Low carbohydrate ketogenic diets[]
The ketone body beta-hydroxybutyrate is a fuel source for the brain during times of fasting when blood glucose levels fall. Although the mechanism is not understood, it is well established that eating a diet low in carbohydrates can be therapeutic for children with epilepsy.[174] This is likely a result of ketone bodies providing an alternative fuel source to glucose for neuronal function. Furthermore, a ketogenic diet can be beneficial for dementia patients.[175] Medium-chain triglycerides can stimulate ketone synthesis[176] and coconut oil is a rich source of medium chain triglycerides that several anecdotal reports suggest can improve cognitive function in Alzheimer's type dementia patients.[177][178]
Protein[]
When protein is consumed, it is broken down into amino acids. These amino acids are used to produce many things like neurotransmitters, enzymes, hormones, and chromosomes. Proteins known as complete proteins contain all eight of the essential amino acids. Meat, cheese, eggs, and yogurt are all examples of complete proteins. Incomplete proteins contain only some of the eight essential amino acids and it is recommended that people consume a combination of these proteins. Examples of incomplete proteins include nuts, seeds, legumes, and grains.[179] When animals are fed a diet deficient in essential amino acids, uncharged tRNAs accumulate in the anterior piriform cortex signaling diet rejection [105]. The body normally interconverts amino acids to maintain homeostasis, but muscle protein can be catabolized to release amino acids during conditions of amino acid deficiency. Disruption of amino acid metabolism can affect brain development and neurophysiology to affect behavior. For example, fetal protein deficiency decreases the number of neurons in the CA1 region of the hippocampus.[180]
Glutamate[]
Glutamate is a proteinogenic amino acid and neurotransmitter, though it is perhaps publicly best known in its sodium salt form: monosodium glutamate (MSG). It is also a flavor on its own, producing the umami or savory flavor found in many fermented foods such as cheese. As an amino acid it acts as a source of fuel for various cellular functions and as a neurotransmitter. Glutamate operates as an excitatory neurotransmitter and is released when a nerve impulse excites a glutamate producing cell. This in turn binds to neurons with glutamate receptors, stimulating them.
Deficiencies and treatments[]
Glutamate is a nutrient that is extremely difficult to be deficient in, as, being an amino acid, it is found in every food that contains protein. Additionally it is found, as previously mentioned, in fermented foods and in foods containing monosodium glutamate. As such, good sources of glutamate include meat, fish, dairy products and a wide array of other foods. Glutamate is also absorbed extremely efficiently by the intestines.[181] However, there are instances of glutamate deficiency occurring, but only in cases where genetic disorders are present. One such example is Glutamate formiminotransferase deficiency and can cause either minor or profound physical and intellectual disabilities, depending on the severity of the condition. This disorder is extremely rare however, as only twenty people have so far been identified with this condition.[182] Glutamate, while critically important in the body also acts as an excitotoxin in high concentrations not normally found outside of laboratory conditions,[183] although it can occur following brain injury or spinal cord injury.[184]
Phenylalanine[]
L-Phenylalanine is biologically converted into L-tyrosine, another one of the DNA-encoded amino acids, and beta-phenethylamine.[185] L-tyrosine in turn is converted into L-DOPA, which is further converted into dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline). The latter three are known as the catecholamines. Phenethylamine is further converted into N-methylphenethylamine.[186] Phenylalanine uses the same active transport channel as tryptophan to cross the blood–brain barrier, and, in large quantities, interferes with the production of serotonin.[187]
Phenylketonuria[]
Toxic levels of phenylalanine accumulate in the brains of patients with phenylketonuria leading to severe brain damage and mental retardation. To prevent brain damage, these individuals can restrict dietary phenylalanine intake by avoiding protein and supplementing their diet with essential amino acids.[188]
See also[]
References[]
- ^ Bedi KS (June 2003). "Nutritional effects on neuron numbers". Nutritional Neuroscience. 6 (3): 141–52. doi:10.1080/1028415031000098549. PMID 12793518.
- ^ Dauncey MJ (November 2009). "New insights into nutrition and cognitive neuroscience". Proceedings of the Nutrition Society. 68 (4): 408–15. doi:10.1017/S0029665109990188. PMID 19698201. S2CID 21999711.
- ^ Fonseca-Azevedo K., Herculano-Houzel S.; Herculano-Houzel (2012). "Metabolic constraint imposes tradeoff between body size and number of brain neurons in human evolution". Proceedings of the National Academy of Sciences. 109 (45): 18571–18576. Bibcode:2012PNAS..10918571F. doi:10.1073/pnas.1206390109. PMC 3494886. PMID 23090991.
- ^ Jump up to: a b Gómez-Pinilla, Fernando (2008). "Brain foods: The effects of nutrients on brain function". Nature Reviews Neuroscience. 9 (7): 568–78. doi:10.1038/nrn2421. PMC 2805706. PMID 18568016.
- ^ Alamy M.; Bengelloun W. A. (2012). "Malnutrition and brain development: an analysis of the effects of inadequate diet during different stages of life in rat". Neuroscience & Biobehavioral Reviews. 36 (6): 1463–1480. doi:10.1016/j.neubiorev.2012.03.009. PMID 22487135. S2CID 207089666.
- ^ Schrag M.; Mueller C.; Oyoyo U.; Smith M. A.; Kirsch W. M. (2011). "Iron, zinc and copper in the Alzheimer's disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion". Progress in Neurobiology. 94 (3): 296–306. doi:10.1016/j.pneurobio.2011.05.001. PMC 3134620. PMID 21600264.
- ^ Duce J. A., Tsatsanis A., Cater M. A., James S. A., Robb E., Wikhe K., Bush A. I.; Tsatsanis; Cater; James; Robb; Wikhe; Leong; Perez; Johanssen; Greenough; Cho; Galatis; Moir; Masters; McLean; Tanzi; Cappai; Barnham; Ciccotosto; Rogers; Bush (2010). "Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer's disease". Cell. 142 (6): 857–867. doi:10.1016/j.cell.2010.08.014. PMC 2943017. PMID 20817278.CS1 maint: multiple names: authors list (link)
- ^ Chang S.; Zeng L.; Brouwer I. D.; Kok F. J.; Yan H. (2013). "Effect of Iron Deficiency Anemia in Pregnancy on Child Mental Development in Rural China". Pediatrics. 131 (3): e755-63. doi:10.1542/peds.2011-3513. PMID 23400604. S2CID 655350.
- ^ Kwik-Uribe Catherine L.; et al. (2000). "Chronic marginal iron intakes during early development in mice result in persistent changes in dopamine metabolism, myelin composition". The Journal of Nutrition. 130 (11): 2821–2830. doi:10.1093/jn/130.11.2821. PMID 11053527.
- ^ Kwik-Uribe Catherine L.; Golub Mari S.; Keen Carl L. (2000). "Chronic marginal iron intakes during early development in mice alter brain iron concentrations and behavior despite postnatal iron supplementation". The Journal of Nutrition. 130 (8): 2040–2048. doi:10.1093/jn/130.8.2040. PMID 10917923.
- ^ Lee Dawn L.; et al. (2012). "Iron deficiency disrupts axon maturation of the developing auditory nerve". The Journal of Neuroscience. 32 (14): 5010–5015. doi:10.1523/jneurosci.0526-12.2012. PMC 3327472. PMID 22492056.
- ^ Golub Mari S.; et al. (2006). "Behavioral consequences of developmental iron deficiency in infant rhesus monkeys". Neurotoxicology and Teratology. 28 (1): 3–17. doi:10.1016/j.ntt.2005.10.005. PMC 1540448. PMID 16343844.
- ^ Golub Mari S.; Hogrefe Casey E.; Unger Erica L. (2012). "Influence of prenatal iron deficiency and MAOA genotype on response to social challenge in rhesus monkey infants". Genes, Brain and Behavior. 11 (3): 278–290. doi:10.1111/j.1601-183x.2012.00772.x. PMC 3511847. PMID 22340208.
- ^ "WHO - Archived: Daily iron and folic acid supplementation in pregnant women". WHO.
- ^ Ojukwu J. U.; Okebe J. U.; Yahav D.; Paul M. (2010). "Cochrane review: Oral iron supplementation for preventing or treating anaemia among children in malaria‐endemic areas". Evidence-Based Child Health: A Cochrane Review Journal. 5 (2): 967–1183. doi:10.1002/ebch.542.
- ^ Maret, Wolfgang (2013). "Chapter 14 Zinc and the Zinc Proteome". In Banci, Lucia (ed.). Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. doi:10.1007/978-94-007-5561-10_14 (inactive 31 May 2021). ISBN 978-94-007-5560-4.CS1 maint: DOI inactive as of May 2021 (link) electronic-book ISBN 978-94-007-5561-1 ISSN 1559-0836 electronic-ISSN 1868-0402
- ^ Saadi R. A., He K., Hartnett K. A., Kandler K., Hershfinkel M., Aizenman E.; He; Hartnett; Kandler; Hershfinkel; Aizenman (2012). "SNARE-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro". Neuroscience. 210: 38–46. doi:10.1016/j.neuroscience.2012.03.001. PMC 3358579. PMID 22441041.CS1 maint: multiple names: authors list (link)
- ^ Dvergsten C. L.; Johnson L. A.; Sandstead H. H. (1984). "Alterations in the postnatal development of the cerebellar cortex due to zinc deficiency. III. Impaired dendritic differentiation of basket and stellate cells". Developmental Brain Research. 16 (1): 21–26. doi:10.1016/0165-3806(84)90058-0. PMID 6488052.
- ^ Jump up to: a b Nuttall J. R.; Oteiza P. I. (2012). "Zinc and the ERK kinases in the developing brain". Neurotoxicity Research. 21 (1): 128–141. doi:10.1007/s12640-011-9291-6. PMC 4316815. PMID 22095091.
- ^ Tassabehji N. M., Corniola R. S., Alshingiti A., Levenson C. W.; Corniola; Alshingiti; Levenson (2008). "Zinc deficiency induces depression-like symptoms in adult rats". Physiology & Behavior. 95 (3): 365–369. doi:10.1016/j.physbeh.2008.06.017. PMID 18655800. S2CID 23430644.CS1 maint: multiple names: authors list (link)
- ^ Doherty K., Connor M., Cruickshank R.; Connor; Cruickshank (2011). "Zinc-containing denture adhesive: a potential source of excess zinc resulting in copper deficiency myelopathy". British Dental Journal. 210 (11): 523–525. doi:10.1038/sj.bdj.2011.428. PMID 21660014. S2CID 9995046.CS1 maint: multiple names: authors list (link)
- ^ Maret W, Sandstead HH; Sandstead (2006). "Zinc requirements and the risks and benefits of zinc supplementation". J Trace Elem Med Biol. 20 (1): 3–18. doi:10.1016/j.jtemb.2006.01.006. PMID 16632171.
- ^ "zinc deficiency". GPnotebook.
- ^ Prasad AS (2003). "Zinc deficiency : Has been known of for 40 years but ignored by global health organisations". BMJ. 326 (7386): 409–10. doi:10.1136/bmj.326.7386.409. PMC 1125304. PMID 12595353.
- ^ El-Safty Ibrahim A M; Gadallah Mohsen; Shafik Ahmed; Shouman Ahmed E (2002). "Effect of mercury vapour exposure on urinary excretion of calcium, zinc and copper: relationship to alterations in functional and structural integrity of the kidney". Toxicol Ind Health. 18 (8): 377–388. doi:10.1191/0748233702th160oa. PMID 15119526. S2CID 32876828.
- ^ Funk Day, Brady (1987). "Displacement of zinc and copper from copper-induced metallothionein by cadmium and by mercury: in vivo and ex vivo studies". Comp Biochem Physiol C. 86 (1): 1–6. doi:10.1016/0742-8413(87)90133-2. PMID 2881702.
- ^ Solomons, N.W. (2001) Dietary Sources of zinc and factors affecting its bioavailability. Food Nutr. Bull. 22: 138-154
- ^ Sandstead HH (1991). "Zinc deficiency. A public health problem?". Am. J. Dis. Child. 145 (8): 853–9. doi:10.1001/archpedi.1991.02160080029016. PMID 1858720.
- ^ Castillo-Duran C, Vial P, Uauy R; Vial; Uauy (1988). "Trace mineral balance during acute diarrhea in infants". J. Pediatr. 113 (3): 452–7. doi:10.1016/S0022-3476(88)80627-9. PMID 3411389.CS1 maint: multiple names: authors list (link)
- ^ Manary MJ, Hotz C, Krebs NF, et al. (2000). "Dietary phytate reduction improves zinc absorption in Malawian children recovering from tuberculosis but not in well children". J. Nutr. 130 (12): 2959–64. doi:10.1093/jn/130.12.2959. PMID 11110854.
- ^ Gibson RS (2006). "Zinc: the missing link in combating micronutrient malnutrition in developing countries". Proc Nutr Soc. 65 (1): 51–60. doi:10.1079/PNS2005474. PMID 16441944. S2CID 3656729.
- ^ Suzuki H, Asakawa A, Li JB, Tsai M, Amitani H, Ohinata K, Komai M, Inui A (September 2011). "Zinc as an appetite stimulator - the possible role of zinc in the progression of diseases such as cachexia and sarcopenia". Recent Patents on Food, Nutrition & Agriculture. 3 (3): 226–31. doi:10.2174/2212798411103030226. PMID 21846317.
- ^ Birmingham C. L.; Goldner E. M.; Bakan R. (1994). "Controlled trial of zinc supplementation in anorexia nervosa". International Journal of Eating Disorders. 15 (3): 251–255. doi:10.1002/1098-108X(199404)15:3<251::AID-EAT2260150308>3.0.CO;2-# (inactive 31 May 2021). PMID 8199605.CS1 maint: DOI inactive as of May 2021 (link)
- ^ Sanstead H. H.; et al. (2000). "Zinc nutriture as related to brain". J. Nutr. 130: 140S–146S.
- ^ Jump up to: a b Black MM (2003). "The Evidence Linking Zinc Deficiency with Children's Cognitive and Motor Functioning". J. Nutr. 133 (5 Suppl 1): 1473S–6S. doi:10.1093/jn/133.5.1473S. PMC 3137935. PMID 12730446.
- ^ Jump up to: a b Black MM (1998). "Zinc deficiency and child development". Am. J. Clin. Nutr. 68 (2 Suppl): 464S–9S. doi:10.1093/ajcn/68.2.464S. PMC 3137936. PMID 9701161.
- ^ Nuttall, J; Oteiza (2012). "Zinc and the ERK kinases in the developing brain". Neurotoxicity Research. 21 (1): 128–141. doi:10.1007/s12640-011-9291-6. PMC 4316815. PMID 22095091.
- ^ Sawada T, Yokoi K; Yokoi (March 2010). "Effect of zinc supplementation on mood states in young women: a pilot study". Eur J Clin Nutr. 64 (3): 331–3. doi:10.1038/ejcn.2009.158. PMID 20087376. S2CID 31660050.
- ^ Nelson K. T., Prohaska J. R.; Prohaska (2009). "Copper deficiency in rodents alters dopamine b-mono-oxygenase activity, mRNA and protein level". British Journal of Nutrition. 102 (1): 18–28. doi:10.1017/S0007114508162961. PMID 19079842. S2CID 25353691.
- ^ Jump up to: a b c d e f g h Jaiser S. R.; Winston G. P. (2010). "Copper deficiency myelopathy". Journal of Neurology. 257 (6): 869–881. doi:10.1007/s00415-010-5511-x. PMC 3691478. PMID 20232210.
- ^ Halfdanarson T. R.; Kumar N.; Li C. Y.; Phyliky R. L.; Hogan W. J. (2008). "Hematological manifestations of copper deficiency: a retrospective review. [Article]". European Journal of Haematology. 80 (6): 523–531. doi:10.1111/j.1600-0609.2008.01050.x. PMID 18284630. S2CID 38534852.
- ^ Kodama H.; Fujisawa C. (2009). "Copper metabolism and inherited copper transport disorders: molecular mechanisms, screening, and treatment". Metallomics. 1 (1): 42–52. doi:10.1039/b816011m.
- ^ Jump up to: a b c d e Kumar N (2006). "Copper deficiency myelopathy (human swayback)". Mayo Clinic Proceedings. 81 (10): 1371–1384. doi:10.4065/81.10.1371. PMID 17036563.
- ^ Jump up to: a b Jaiser, S. R., & Winston, G. P. (2008). Copper deficiency myelopathy and subacute combined degeneration of the cord: why is the phenotype so similar?" Journal of Neurology 255, P569.
- ^ Ataxic Gait Demonstration. Online Medical Video. https://www.youtube.com/watch?v=FpiEprzObIU
- ^ Jump up to: a b Bolamperti L.; Leone M. A.; Stecco A.; Reggiani M.; Pirisi M.; Carriero A.; et al. (2009). "Myeloneuropathy due to copper deficiency: clinical and MRI findings after copper supplementation. [Article]". Neurological Sciences. 30 (6): 521–524. doi:10.1007/s10072-009-0126-7. PMID 19768378. S2CID 21488713.
- ^ Jump up to: a b c d e f Pineles S. L.; Wilson C. A.; Balcer L. J.; Slater R.; Galetta S. L. (2010). "Combined Optic Neuropathy and Myelopathy Secondary to Copper Deficiency. [Review]". Survey of Ophthalmology. 55 (4): 386–392. doi:10.1016/j.survophthal.2010.02.002. PMID 20451943.
- ^ Jump up to: a b Jaiser, Stephan R. and Duddy, R. Copper Deficiency Masquerading as Subacute Combined Degeneration of the Cord and Myelodysplastic Syndrome. Advances in clinical neuroscience and rehabilitation, http://www.acnr.co.uk/JA07/ACNR_JA07_abnwinner.pdf
- ^ Spinazzi M.; De Lazzari F.; Tavolato B.; Angelini C.; Manara R.; Armani M. (2007). "Myelo-optico-neuropathy in copper deficiency occurring after partial gastrectomy. Do small bowel bacterial overgrowth syndrome and occult zinc ingestion tip the balance?". Journal of Neurology. 254 (8): 1012–1017. doi:10.1007/s00415-006-0479-2. PMID 17415508. S2CID 28373986.
- ^ Held KD; et al. (May 1996). "Role of Fenton chemistry in thiol-induced toxicity and apoptosis". Radiat. Res. Radiation Research Society. 145 (5): 542–53. Bibcode:1996RadR..145..542H. doi:10.2307/3579272. JSTOR 3579272. PMID 8619019.
- ^ Brewer GJ (February 2007). "Iron and copper toxicity in diseases of aging, particularly atherosclerosis and Alzheimer's disease". Exp. Biol. Med. (Maywood). 232 (2): 323–35. PMID 17259340.
- ^ Wolf T. L.; Kotun J.; Meador-Woodruff J. H. (2006). "Plasma copper, iron, ceruloplasmin and ferroxidase activity in schizophrenia". Schizophrenia Research. 86 (1): 167–171. doi:10.1016/j.schres.2006.05.027. PMID 16842975. S2CID 38267889.
- ^ Brewer GJ (April 2010). "Copper toxicity in the general population". Clin Neurophysiol. 121 (4): 459–60. doi:10.1016/j.clinph.2009.12.015. PMID 20071223. S2CID 43106197.
- ^ Faller P (14 December 2009). "Copper and zinc binding to amyloid-beta: coordination, dynamics, aggregation, reactivity and metal-ion transfer". ChemBioChem. 10 (18): 2837–45. doi:10.1002/cbic.200900321. PMID 19877000. S2CID 35130040.
- ^ Hureau C, Faller P; Faller (October 2009). "Abeta-mediated ROS production by Cu ions: structural insights, mechanisms and relevance to Alzheimer's disease". Biochimie. 91 (10): 1212–7. doi:10.1016/j.biochi.2009.03.013. PMID 19332103.
- ^ Brewer G. J. (2012). "Copper excess, zinc deficiency, and cognition loss in Alzheimer's disease". BioFactors. 38 (2): 107–113. doi:10.1002/biof.1005. PMID 22438177. S2CID 16989047.
- ^ Food and Nutrition Board, Institute of Medicine. Manganese. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:394-419. (National Academy Press)
- ^ Keen CL, Zidenberg-Cherr S. Manganese. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:334-343.
- ^ Jump up to: a b Roth JA (2006). "Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination". Biol. Res. 39 (1): 45–57. doi:10.4067/S0716-97602006000100006. PMID 16629164.
- ^ Couper, J. (1837). "Sur les effets du peroxide de manganèse". Journal de chimie médicale, de pharmacie et de toxicologie. 3: 223–225.
- ^ Jump up to: a b Yin, Zhaobao; Jiang, Haiyan; Lee, Eun-Sook Y.; Ni, Mingwei; Erikson, Keith M.; Milatovic, Dejan; Bowman, Aaron B.; Aschner, Michael (2010). "Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation" (PDF). Journal of Neurochemistry. 112 (5): 1190–8. doi:10.1111/j.1471-4159.2009.06534.x. PMC 2819584. PMID 20002294.
- ^ Fryzek JP, Hansen J, Cohen S, Bonde JP, Llambias MT, Kolstad HA, Skytthe A, Lipworth L, Blot WJ, Olsen JH (May 2005). "A cohort study of Parkinson's disease and other neurodegenerative disorders in Danish welders" (PDF). Journal of Occupational and Environmental Medicine. 47 (5): 466–72. doi:10.1097/01.jom.0000161730.25913.bf. PMID 15891525. S2CID 29870690.
- ^ Fored, C M; Fryzek, JP; Brandt, L; Nise, G; Sjögren, B; McLaughlin, JK; Blot, WJ; Ekbom, A (2006). "Parkinson's disease and other basal ganglia or movement disorders in a large nationwide cohort of Swedish welders". Occupational and Environmental Medicine. 63 (2): 135–40. doi:10.1136/oem.2005.022921. PMC 2078076. PMID 16421393.
- ^ Marsh GM, Gula MJ; Gula (October 2006). "Employment as a welder and Parkinson disease among heavy equipment manufacturing workers". Journal of Occupational and Environmental Medicine. 48 (10): 1031–46. doi:10.1097/01.jom.0000232547.74802.d8. PMID 17033503. S2CID 1355456.
- ^ de Bie RM, Gladstone RM, Strafella AP, Ko JH, Lang AE; Gladstone; Strafella; Ko; Lang (June 2007). "Manganese-induced Parkinsonism associated with methcathinone (Ephedrone) abuse". Arch. Neurol. 64 (6): 886–9. doi:10.1001/archneur.64.6.886. PMID 17562938.CS1 maint: multiple names: authors list (link)
- ^ Sanotsky, Y., Lesyk, R., Fedoryshyn, L., Komnatska, I., Matviyenko, Y. and Fahn, S. (June 2007). "Manganic encephalopathy due to "ephedrone" abuse". Movement Disorders. 22 (9): 1337–1343. doi:10.1002/mds.21378. PMID 17566121. S2CID 11564105.CS1 maint: multiple names: authors list (link)
- ^ Ensing, J. G. (1985). "Bazooka: Cocaine-Base and Manganese Carbonate". Journal of Analytical Toxicology. 9 (1): 45–46. doi:10.1093/jat/9.1.45. PMID 3981975.
- ^ Kondakis, Xenophon G.; Makris, Nicolas; Leotsinidis, Michael; Prinou, Mary; Papapetropoulos, Theodore (1989). "Possible Health Effects of High Manganese Concentration in Drinking Water". Archives of Environmental Health. 44 (3): 175–178. doi:10.1080/00039896.1989.9935883. PMID 2751354.
- ^ Hudnell, HK (1999). "Effects from environmental Mn exposures: A review of the evidence from non-occupational exposure studies". Neurotoxicology. 20 (2–3): 379–397. PMID 10385898.
- ^ Lynam, DR; Roos, JW; Pfeifer, GD; Fort, BF; Pullin, TG (1999). "Environmental effects and exposures to manganese from use of methylcyclopentadienyl manganese tricarbonyl (MMT) in gasoline". Neurotoxicology. 20 (2–3): 145–150. PMID 10385878.
- ^ Reynolds JG, Roos JW, Wong J, Deutsch SE. Manganese particulates from vehicles using MMT fuel. In 15th International Neurotoxicology Conference, Little Rock, AR, 1997.
- ^ Lynam, D.R.; Pfeifer, G.D.; Fort, B.F.; Gelbcke, A.A. (1990). "Environmental assessment of MMT™ fuel additive". Science of the Total Environment. 93: 107–114. Bibcode:1990ScTEn..93..107L. doi:10.1016/0048-9697(90)90098-F. PMID 2113712.
- ^ Ferraz, H. B.; f. Bertolucci, P. H.; Pereira, J. S.; Lima, J.G.C.; f. Andrade, L. A. (1988). "Chronic exposure to the fungicide maneb may produce symptoms and signs of CNS manganese intoxication". Neurology. 38 (4): 550–553. doi:10.1212/WNL.38.4.550. PMID 3352909. S2CID 20400188.
- ^ Ballatori N. Molecular mechanisms of hepatic metal transport. In Molecular Biology and Toxicology of Metals, Zalups RK, Koropatnick J (eds). Taylor & Francis: New York, 2000; 346-381.
- ^ Jump up to: a b Verity, MA (1999). "Manganese neurotoxicity: A mechanistic hypothesis". Neurotoxicology. 20 (2–3): 489–497. PMID 10385907.
- ^ Zheng, Wei; Zhao, Qiuqu (2001). "Iron overload following manganese exposure in cultured neuronal, but not neuroglial cells". Brain Research. 897 (1–2): 175–179. doi:10.1016/S0006-8993(01)02049-2. PMC 3980869. PMID 11282372.
- ^ Zheng, Wei; Zhao, Qiuqu; Slavkovich, Vesna; Aschner, Michael; Graziano, Joseph H (1999). "Alteration of iron homeostasis following chronic exposure to manganese in rats". Brain Research. 833 (1): 125–132. doi:10.1016/S0006-8993(99)01558-9. PMC 4126166. PMID 10375687.
- ^ Zheng, Wei (2001). "Neurotoxicology of the Brain Barrier System: New Implications" (PDF). Clinical Toxicology. 39 (7): 711–719. doi:10.1081/CLT-100108512. PMC 4111935. PMID 11778669.
- ^ Jump up to: a b c Lai, JC; Minski, MJ; Chan, AW; Leung, TK; Lim, L (1999). "Manganese mineral interactions in brain". Neurotoxicology. 20 (2–3): 433–444. PMID 10385902.
- ^ Zheng, Wei; Ren, Sean; Graziano, Joseph H. (1998). "Manganese inhibits mitochondrial aconitase: A mechanism of manganese neurotoxicity". Brain Research. 799 (2): 334–342. doi:10.1016/S0006-8993(98)00481-8. PMC 4126159. PMID 9675333.
- ^ Li G, Zhang L, Lu L, Wu P, Zheng W (2004). "Occupational exposure to welding fume among welders: alterations of manganese, iron, zinc, copper, and lead in body fluids and the oxidative stress status". J. Occup. Environ. Med. 46 (3): 241–248. doi:10.1097/01.jom.0000116900.49159.03. PMC 4126160. PMID 15091287.
- ^ Lauwerys, Robert; et al. (1985). "Fertility of male workers exposed to mercury vapor or to manganese dust: A questionnaire study". American Journal of Industrial Medicine. 7 (2): 171–176. doi:10.1002/ajim.4700070208. PMID 3976664.
- ^ Treinen, Kimberley A.; Gray, Tim J. B.; Blazak, William F. (1995). "Developmental toxicity of mangafodipir trisodium and manganese chloride in Sprague-Dawley rats". Teratology. 52 (2): 109–115. doi:10.1002/tera.1420520207. PMID 8588182.
- ^ Donaldson J, McGregor D, LaBella F (1982). "Manganese neurotoxicity: a model for free radical mediated neurodegeneration?". Can. J. Physiol. Pharmacol. 60 (11): 1398–405. doi:10.1139/y82-208. PMID 6129921.
- ^ Lee, J.-W. (2000). "Manganese Intoxication". Archives of Neurology. 57 (4): 597–599. doi:10.1001/archneur.57.4.597. PMID 10768639.
- ^ Mena I, Court J, Fuenzalida S, Papavasiliou PS, Cotzias GC. Modification of chronic manganese poisoning. Treatment with Ldopa or 5-OH tryptophane. New Engl. J. Med. 1970; 282(1) 5-10.
- ^ Rosenstock, Harvey A. (1971). "Chronic Manganism: Neurologic and Laboratory Studies During Treatment with Levodopa". JAMA: The Journal of the American Medical Association. 217 (10): 1354–1358. doi:10.1001/jama.1971.03190100038007.
- ^ Huang, C.-C.; Lu, C.-S.; Chu, N.-S.; Hochberg, F.; Lilienfeld, D.; Olanow, W.; Calne, D. B. (1993). "Progression after chronic manganese exposure". Neurology. 43 (8): 1479–1483. doi:10.1212/WNL.43.8.1479. PMID 8351000. S2CID 12621501.
- ^ Huang, C.-C.; Chu, N.-S.; Lu, C.-S.; Chen, R.-S.; Calne, D. B. (1998). "Long-term progression in chronic manganism: Ten years of follow-up". Neurology. 50 (3): 698–700. doi:10.1212/WNL.50.3.698. PMID 9521259. S2CID 34347615.
- ^ Ono, Kenjiro; Komai, Kiyonobu; Yamada, Masahito (2002). "Myoclonic involuntary movement associated with chronic manganese poisoning". Journal of the Neurological Sciences. 199 (1–2): 93–96. doi:10.1016/S0022-510X(02)00111-9. PMID 12084450. S2CID 40327454.
- ^ Calne, DB; Chu, NS; Huang, CC; Lu, CS; Olanow, W (1994). "Manganism and idiopathic parkinsonism: Similarities and differences". Neurology. 44 (9): 1583–1586. doi:10.1212/WNL.44.9.1583. PMID 7936278. S2CID 28563940.
- ^ "Study: Arsenic contaminates 27 percent of wells in Vietnam's Red Rive…".
- ^ Nowak, L., Bregestovski, P., Ascher, P., Herbet, A., & Prochiantz, A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones.
- ^ Pan H. C.; Sheu M. L.; Su H. L.; Chen Y. J.; Chen C. J.; Yang D. Y.; Cheng F. C. (2011). "Magnesium supplement promotes sciatic nerve regeneration and down-regulates inflammatory response". Magnesium Research. 24 (2): 54–70. doi:10.1684/mrh.2011.0280. PMID 21609904.
- ^ FDA DRI Report on Magnesium http://www.nal.usda.gov/fnic/DRI//DRI_Calcium/190-249.pdf Archived 17 July 2013 at the Wayback Machine
- ^ McCann, J. C., & Ames, B. N. (2010). 24 Evidence Required for Causal Inferences about Effects of Micronutrient Deficiencies during Development on Brain Health. Micronutrients and Brain Health, 355.
- ^ Ames B. N. (2006). "Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage". Proceedings of the National Academy of Sciences. 103 (47): 17589–17594. Bibcode:2006PNAS..10317589A. doi:10.1073/pnas.0608757103. PMC 1693790. PMID 17101959.
- ^ Jump up to: a b Li, T.-Y.; Huang, H.-M.; Mao, C.-T.; Liu, Y.; Qu, P.; Yang, L. (2008). "Marginal Vitamin a Deficiency in Pregnancy Can Induce Memory Deficit in Adult Offspring". Pediatrics. 121: S152.2–S153. doi:10.1542/peds.2007-2022NNNNNN. S2CID 72007116.
- ^ Li, T.-Y.; Mao, C.-T.; Huang, H.-M.; Liu, Y.-X.; Qu, P.; Yang, L. (2008). "Effects of Marginal Vitamin a Deficiency on Long-Term Potentiation in Young Rats". Pediatrics. 121: S153.1–S153. doi:10.1542/peds.2007-2022OOOOOO. S2CID 51762665.
- ^ Jump up to: a b Etchamendy, Nicole; Enderlin, Valérie; Marighetto, Aline; Pallet, Véronique; Higueret, Paul; Jaffard, Robert (2003). "Vitamin a deficiency and relational memory deficit in adult mice: Relationships with changes in brain retinoid signalling". Behavioural Brain Research. 145 (1–2): 37–49. doi:10.1016/S0166-4328(03)00099-8. PMID 14529804. S2CID 22724266.
- ^ Jump up to: a b Cocco, S; Diaz, G; Stancampiano, R; Diana, A; Carta, M; Curreli, R; Sarais, L; Fadda, F (2002). "Vitamin a deficiency produces spatial learning and memory impairment in rats". Neuroscience. 115 (2): 475–82. doi:10.1016/S0306-4522(02)00423-2. PMID 12421614. S2CID 14285781.
- ^ Hernández-Pinto, A.M.; Puebla-Jiménez, L.; Arilla-Ferreiro, E. (2006). "A vitamin A-free diet results in impairment of the rat hippocampal somatostatinergic system". Neuroscience. 141 (2): 851–61. doi:10.1016/j.neuroscience.2006.04.034. hdl:10017/2297. PMID 16757122. S2CID 16540757.
- ^ Kheirvari, Sorayya; Uezu, Kayoko; Sakai, Tohru; Nakamori, Masayo; Alizadeh, Mohammad; Sarukura, Nobuko; Yamamoto, Shigeru (2006). "Increased Nerve Growth Factor by Zinc Supplementation with Concurrent Vitamin a Deficiency Does Not Improve Memory Performance in Mice". Journal of Nutritional Science and Vitaminology. 52 (6): 421–7. doi:10.3177/jnsv.52.421. PMID 17330505.
- ^ Jump up to: a b c Ogershok, Paul R.; Rahman, Aamer; Nestor, Scott; Brick, James (2002). "Wernicke Encephalopathy in Nonalcoholic Patients". American Journal of the Medical Sciences. 323 (2): 107–11. doi:10.1097/00000441-200202000-00010. PMID 11863078. S2CID 12996092.
- ^ Bourre, JM (2006). "Effects of nutrients (in food) on the structure and function of the nervous system: Update on dietary requirements for brain. Part 1: Micronutrients". The Journal of Nutrition, Health & Aging. 10 (5): 377–85. PMID 17066209.
- ^ Thompson, J (2005). "Vitamins, minerals and supplements: Part two". Community Practitioner. 78 (10): 366–8. PMID 16245676.
- ^ Jump up to: a b c d e Bond, Nigel W.; Homewood, Judi (1991). "Wernicke's encephalopathy and Korsakoff's psychosis: To fortify or not to fortify?". Neurotoxicology and Teratology. 13 (4): 353–5. doi:10.1016/0892-0362(91)90083-9. PMID 1921914.
- ^ Jump up to: a b c Cooper, Jack R.; Pincus, Jonathan H. (1979). "The role of thiamine in nervous tissue". Neurochemical Research. 4 (2): 223–39. doi:10.1007/BF00964146. PMID 37452. S2CID 22390486.
- ^ Bâ A (2011). "Comparative effects of alcohol and thiamine deficiency on the developing central nervous system". Behavioural Brain Research. 225 (1): 235–242. doi:10.1016/j.bbr.2011.07.015. PMID 21784107. S2CID 32144686.
- ^ Jump up to: a b c Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline (PDF). Washington, DC: National Academy Press. 1998. ISBN 0-309-06554-2. Archived from the original (PDF) on 18 June 2013. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Richardson S.; Malhotra A.; Cwynarski K.; Hughes D.; Prentice A.; McNamara C. (2010). "Patients undergoing high dose chemotherapy for primary CNS lymphoma should receive prophylactic thiamine to prevent Wernike's encephalopathy". British Journal of Haematology. 149 (6): 899–901. doi:10.1111/j.1365-2141.2010.08112.x. PMID 20151977. S2CID 8014387.
- ^ Jump up to: a b c d e f Singleton., C.K.; Martin, P.R. (2001). "Molecular Mechanisms of Thiamine Utilization". Current Molecular Medicine. 1 (2): 197–207. doi:10.2174/1566524013363870. PMID 11899071.
- ^ Pitel A. L., Zahr N. M., Jackson K., Sassoon S. A., Rosenbloom M. J., Pfefferbaum A., Sullivan E. V.; Zahr; Jackson; Sassoon; Rosenbloom; Pfefferbaum; Sullivan (2010). "Signs of preclinical Wernicke's encephalopathy and thiamine levels as predictors of neuropsychological deficits in alcoholism without Korsakoff's syndrome". Neuropsychopharmacology. 36 (3): 580–588. doi:10.1038/npp.2010.189. PMC 3055684. PMID 20962766.CS1 maint: multiple names: authors list (link)
- ^ Hoyumpa, Anastacio M. (1983). "Alcohol and Thiamine Metabolism". Alcoholism: Clinical and Experimental Research. 7 (1): 11–14. doi:10.1111/j.1530-0277.1983.tb05403.x. PMID 6342440.
- ^ Jump up to: a b c Harper, C (1979). "Wernicke's encephalopathy: A more common disease than realised. A neuropathological study of 51 cases". Journal of Neurology, Neurosurgery & Psychiatry. 42 (3): 226–31. doi:10.1136/jnnp.42.3.226. PMC 490724. PMID 438830.
- ^ Jump up to: a b Harper, C G; Giles, M; Finlay-Jones, R (1986). "Clinical signs in the Wernicke-Korsakoff complex: A retrospective analysis of 131 cases diagnosed at necropsy". Journal of Neurology, Neurosurgery & Psychiatry. 49 (4): 341–5. doi:10.1136/jnnp.49.4.341. PMC 1028756. PMID 3701343.
- ^ Vitamin Basics: The facts about Vitamins in Nutrition (PDF). Germany: DSM Nutritional Products Ltd. 2007. Archived from the original (PDF) on 25 February 2008. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Jump up to: a b c d e Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline (PDF). Washington, DC: National Academy Press. 1998. ISBN 0-309-06554-2. Archived from the original (PDF) on 4 March 2012. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Yehuda, Shlomo; Rabinovitz, Sharon; Mostofsky, David I. (1999). "Essential fatty acids are mediators of brain biochemistry and cognitive functions". Journal of Neuroscience Research. 56 (6): 565–70. doi:10.1002/(SICI)1097-4547(19990615)56:6<565::AID-JNR2>3.0.CO;2-H. PMID 10374811.
- ^ Jump up to: a b c d e f Hegyi, Juraj; Schwartz, Robert A.; Hegyi, Vladimir (2004). "Pellagra: Dermatitis, dementia, and diarrhea". International Journal of Dermatology. 43 (1): 1–5. doi:10.1111/j.1365-4632.2004.01959.x. PMID 14693013. S2CID 33877664.
- ^ Jump up to: a b Flicker, Leon; Ames, David (2005). "Metabolic and endocrinological causes of dementia". International Psychogeriatrics. 17: S79–92. doi:10.1017/S1041610205001961. PMID 16240485. S2CID 2304240.
- ^ Zimmerman, HM (1939). "The Pathology of the Nervous System in Vitamin Deficiencies". The Yale Journal of Biology and Medicine. 12 (1): 23–28.7. PMC 2602501. PMID 21433862.
- ^ Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline (PDF). Washington, DC: National Academy Press. 1998. ISBN 0-309-06554-2. Archived from the original (PDF) on 4 March 2012. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Rainer, M.; Kraxberger, E.; Haushofer, M.; Mucke, H. A. M.; Jellinger, K. A. (2000). "No evidence for cognitive improvement from oral nicotinamide adenine dinucleotide (NADH) in dementia". Journal of Neural Transmission. 107 (12): 1475–81. doi:10.1007/s007020070011. PMID 11459000. S2CID 22789552.
- ^ Morris, M C (2004). "Dietary niacin and the risk of incident Alzheimer's disease and of cognitive decline". Journal of Neurology, Neurosurgery & Psychiatry. 75 (8): 1093–1099. doi:10.1136/jnnp.2003.025858. PMC 1739176. PMID 15258207.
- ^ Milunsky A.; Jick H.; Jick S. S.; Bruell C. L.; MacLaughlin D. S.; Rothman K. J.; Willett W. (1989). "Multivitamin/folic acid supplementation in early pregnancy reduces the prevalence of neural tube defects". JAMA. 262 (20): 2847–2852. doi:10.1001/jama.262.20.2847. PMID 2478730.
- ^ Schmidt R. J.; Tancredi D. J.; Ozonoff S.; Hansen R. L.; Hartiala J.; Allayee H.; Hertz-Picciotto I. (2012). "Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study". The American Journal of Clinical Nutrition. 96 (1): 80–89. doi:10.3945/ajcn.110.004416. PMC 3374734. PMID 22648721.
- ^ Blasko I.; Hinterberger M.; Kemmler G.; Jungwirth S.; Krampla W.; Leitha T.; Fischer P. (2012). "Conversion from mild cognitive impairment to dementia: influence of folic acid and vitamin B12 use in the VITA cohort". The Journal of Nutrition, Health & Aging. 16 (8): 687–694. doi:10.1007/s12603-012-0051-y. PMID 23076510. S2CID 207434580.
- ^ Titaley C. R.; Dibley M. J.; Roberts C. L.; Agho K. (2010). "Combined iron/folic acid supplements and malaria prophylaxis reduce neonatal mortality in 19 sub-Saharan African countries". The American Journal of Clinical Nutrition. 92 (1): 235–243. doi:10.3945/ajcn.2009.29093. PMID 20504976.
- ^ Jump up to: a b c d e f g h i j Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline (PDF). Washington, DC: National Academy Press. 1998. ISBN 0-309-06554-2. Archived from the original (PDF) on 15 May 2013. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Jump up to: a b Hauck, MR (1991). "Cognitive abilities of preschool children: Implications for nurses working with young children". Journal of Pediatric Nursing. 6 (4): 230–5. PMID 1865312.
- ^ Jump up to: a b c Moyers, S; Bailey, LB (2001). "Fetal malformations and folate metabolism: Review of recent evidence". Nutrition Reviews. 59 (7): 215–24. doi:10.1111/j.1753-4887.2001.tb07013.x. PMID 11475447.
- ^ Jump up to: a b Homocysteine Lowering Trialists' Collaboration (2005). "Dose-dependent effects of folic acid on blood concentrations of homocysteine: A meta-analysis of the randomized trials". The American Journal of Clinical Nutrition. 82 (4): 806–12. doi:10.1093/ajcn/82.4.806. PMID 16210710.
- ^ Frye, R. E.; Sequeira, J. M.; Quadros, E. V.; James, S. J.; Rossignol, D. A. (1 March 2013). "Cerebral folate receptor autoantibodies in autism spectrum disorder". Molecular Psychiatry. 18 (3): 369–381. doi:10.1038/mp.2011.175. PMC 3578948. PMID 22230883.
- ^ Jump up to: a b Malouf, Reem; Grimley Evans, John (8 October 2008). "Folic acid with or without vitamin B12 for the prevention and treatment of healthy elderly and demented people". The Cochrane Database of Systematic Reviews (4): CD004514. doi:10.1002/14651858.CD004514.pub2. ISSN 1469-493X. PMID 18843658.
- ^ Jump up to: a b c Bottiglieri, T (1996). "Folate, vitamin B12, and neuropsychiatric disorders". Nutrition Reviews. 54 (12): 382–90. doi:10.1111/j.1753-4887.1996.tb03851.x. PMID 9155210.
- ^ Jump up to: a b Botto, Lorenzo D.; Moore, Cynthia A.; Khoury, Muin J.; Erickson, J. David (1999). "Neural-Tube Defects". New England Journal of Medicine. 341 (20): 1509–19. doi:10.1056/NEJM199911113412006. PMID 10559453.
- ^ Quadri, P; Fragiacomo, C; Pezzati, R; Zanda, E; Forloni, G; Tettamanti, M; Lucca, U (2004). "Homocysteine, folate, and vitamin B-12 in mild cognitive impairment, Alzheimer disease, and vascular dementia". The American Journal of Clinical Nutrition. 80 (1): 114–22. doi:10.1093/ajcn/80.1.114 (inactive 31 May 2021). PMID 15213037.CS1 maint: DOI inactive as of May 2021 (link)
- ^ Shorvon, S D; Carney, M W; Chanarin, I; Reynolds, E H (1980). "The neuropsychiatry of megaloblastic anaemia". BMJ. 281 (6247): 1036–8. doi:10.1136/bmj.281.6247.1036. PMC 1714413. PMID 6253016.
- ^ Bryan, J; Calvaresi, E; Hughes, D (2002). "Short-term folate, vitamin B-12 or vitamin B-6 supplementation slightly affects memory performance but not mood in women of various ages". The Journal of Nutrition. 132 (6): 1345–56. doi:10.1093/jn/132.6.1345. PMID 12042457.
- ^ Wahlin, Åke; Hill, Robert D.; Winblad, Bengt; Bäckman, Lars (1996). "Effects of serum vitamin B-sub-1-sub-2 and folate status on episodic memory performance in very old age: A population-based study". Psychology and Aging. 11 (3): 487–96. doi:10.1037/0882-7974.11.3.487. PMID 8893317.
- ^ Durga, Jane; Van Boxtel, Martin PJ; Schouten, Evert G; Kok, Frans J; Jolles, Jelle; Katan, Martijn B; Verhoef, Petra (2007). "Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: A randomised, double blind, controlled trial". The Lancet. 369 (9557): 208–216. doi:10.1016/S0140-6736(07)60109-3. PMID 17240287. S2CID 20395823.
- ^ Jump up to: a b Fioravanti, M; Ferrario, E; Massaia, M; Cappa, G; Rivolta, G; Grossi, E; Buckley, AE (1998). "Low folate levels in the cognitive decline of elderly patients and the efficacy of folate as a treatment for improving memory deficits". Archives of Gerontology and Geriatrics. 26 (1): 1–13. doi:10.1016/s0167-4943(97)00028-9. PMID 18653121.
- ^ Subar, AF; Block, G; James, LD (1989). "Folate intake and food sources in the US population". The American Journal of Clinical Nutrition. 50 (3): 508–16. doi:10.1093/ajcn/50.3.508. PMID 2773830.
- ^ Zeisel, SH (2004). "Nutritional importance of choline for brain development". Journal of the American College of Nutrition. 23 (6 Suppl): 621S–626S. doi:10.1080/07315724.2004.10719433. PMID 15640516. S2CID 23103875.
- ^ Zeisel S. H. (2006). "The fetal origins of memory: the role of dietary choline in optimal brain development". The Journal of Pediatrics. 149 (5): S131–S136. doi:10.1016/j.jpeds.2006.06.065. PMC 2430654. PMID 17212955.
- ^ Craciunescu C. N.; Johnson A. R.; Zeisel S. H. (2010). "Dietary choline reverses some, but not all, effects of folate deficiency on neurogenesis and apoptosis in fetal mouse brain". The Journal of Nutrition. 140 (6): 1162–1166. doi:10.3945/jn.110.122044. PMC 2869500. PMID 20392884.
- ^ Blusztajn, J.; Wurtman, R. (1983). "Choline and cholinergic neurons". Science. 221 (4611): 614–20. Bibcode:1983Sci...221..614B. doi:10.1126/science.6867732. PMID 6867732. S2CID 8217400.
- ^ Ladd, Sandra L.; Sommer, Susan A.; Laberge, Stephen; Toscano, William (1993). "Brief Report". Clinical Neuropharmacology. 16 (6): 540–9. doi:10.1097/00002826-199312000-00007. PMID 9377589.
- ^ Jump up to: a b c Zeisel, Steven H. (2006). "Choline: Critical Role During Fetal Development and Dietary Requirements in Adults". Annual Review of Nutrition. 26: 229–50. doi:10.1146/annurev.nutr.26.061505.111156. PMC 2441939. PMID 16848706.
- ^ Jump up to: a b Zeisel, Steven H; Da Costa, Kerry-Ann (2009). "Choline: An essential nutrient for public health". Nutrition Reviews. 67 (11): 615–23. doi:10.1111/j.1753-4887.2009.00246.x. PMC 2782876. PMID 19906248.
- ^ Noga, A. A.; Vance, DE (2003). "A Gender-specific Role for Phosphatidylethanolamine N-Methyltransferase-derived Phosphatidylcholine in the Regulation of Plasma High Density and Very Low Density Lipoproteins in Mice". Journal of Biological Chemistry. 278 (24): 21851–9. doi:10.1074/jbc.M301982200. PMID 12668679. S2CID 30737346.
- ^ "Dietary Reference Intakes". Institute of Medicine. Archived from the original on 19 April 2010.
- ^ Alvarez, XA; Laredo, M; Corzo, D; Fernández-Novoa, L; Mouzo, R; Perea, JE; Daniele, D; Cacabelos, R (1997). "Citicoline improves memory performance in elderly subjects". Methods and Findings in Experimental and Clinical Pharmacology. 19 (3): 201–10. PMID 9203170.
- ^ Jump up to: a b c Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline (PDF). Washington, DC: National Academy Press. 1998. ISBN 0-309-06554-2. Archived from the original (PDF) on 12 September 2012. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Jump up to: a b c Vitamin Basics: The facts about Vitamins in Nutrition (PDF). DSM Nutritional Products. 2007. Archived from the original (PDF) on 25 February 2008. Retrieved 2012. Check date values in:
|access-date=(help)[page needed] - ^ Jump up to: a b c d e Baik, H.W.; Russell, R.M. (1999). "Vitamin B12 Deficiency in the Elderly". Annual Review of Nutrition. 19: 357–77. doi:10.1146/annurev.nutr.19.1.357. PMID 10448529.
- ^ Schenk, BE; Kuipers, EJ; Klinkenberg-Knol, EC; Bloemena, EC; Sandell, M; Nelis, GF; Snel, P; Festen, HP; Meuwissen, SG (1999). "Atrophic gastritis during long-term omeprazole therapy affects serum vitamin B12 levels". Alimentary Pharmacology and Therapeutics. 13 (10): 1343–6. doi:10.1046/j.1365-2036.1999.00616.x. PMID 10540050. S2CID 43190262.
- ^ Jump up to: a b c d e Hvas, AM; Nexo, E (2006). "Diagnosis and treatment of vitamin B12 deficiency--an update". Haematologica. 91 (11): 1506–12. PMID 17043022.
- ^ Jump up to: a b c d e f Epstein, Franklin H.; Toh, Ban-Hock; Van Driel, Ian R.; Gleeson, Paul A. (1997). "Pernicious Anemia". New England Journal of Medicine. 337 (20): 1441–8. doi:10.1056/NEJM199711133372007. PMID 9358143.
- ^ Jump up to: a b c Stabler, Sally P.; Allen, Robert H. (2004). "Vitamin B12 Deficiency As a Worldwide Problem". Annual Review of Nutrition. 24: 299–326. doi:10.1146/annurev.nutr.24.012003.132440. PMID 15189123.
- ^ Jump up to: a b Holmes, J. M. (1956). "Cerebral Manifestations of Vitamin-B12 Deficiency". BMJ. 2 (5006): 1394–8. doi:10.1136/bmj.2.5006.1394. PMC 2035923. PMID 13374343.
- ^ Sanders, T. A. B.; Ellis, F. R.; Dickerson, J. W. T. (2007). "Haematological studies on vegans". British Journal of Nutrition. 40 (1): 9–15. doi:10.1079/BJN19780089. PMID 667007.
- ^ Eyles D. W.; Smith S.; Kinobe R.; Hewison M.; McGrath J. J. (2005). "Distribution of the vitamin D receptor and 1α-hydroxylase in human brain". Journal of Chemical Neuroanatomy. 29 (1): 21–30. doi:10.1016/j.jchemneu.2004.08.006. PMID 15589699. S2CID 54334971.
- ^ Kesby J. P., Cui X., O'Loan J., McGrath J. J., Burne T. H., Eyles D. W.; Cui; O'Loan; McGrath; Burne; Eyles (2010). "Developmental vitamin D deficiency alters dopamine-mediated behaviors and dopamine transporter function in adult female rats". Psychopharmacology. 208 (1): 159–168. doi:10.1007/s00213-009-1717-y. PMID 19921153. S2CID 20694610.CS1 maint: multiple names: authors list (link)
- ^ Kesby J. P.; Burne T. H.; McGrath J. J.; Eyles D. W. (2006). "Developmental vitamin D deficiency alters MK 801-induced hyperlocomotion in the adult rat: An animal model of schizophrenia". Biological Psychiatry. 60 (6): 591–596. doi:10.1016/j.biopsych.2006.02.033. PMID 16697353. S2CID 9109008.
- ^ McGrath J., Saari K., Hakko H., Jokelainen J., Jones P., Järvelin M. R., Isohanni M.; Saari; Hakko; Jokelainen; Jones; Järvelin; Chant; Isohanni (2004). "Vitamin D supplementation during the first year of life and risk of schizophrenia: a Finnish birth cohort study". Schizophrenia Research. 67 (2): 237–245. doi:10.1016/j.schres.2003.08.005. PMID 14984883. S2CID 11900260.CS1 maint: multiple names: authors list (link)
- ^ Jump up to: a b Lafourcade M.; Larrieu T.; Mato S.; Duffaud A.; Sepers M.; Matias I.; Manzoni O. J. (2011). "Nutritional omega-3 deficiency abolishes endocannabinoid-mediated neuronal functions" (PDF). Nature Neuroscience. 14 (3): 345–350. doi:10.1038/nn.2736. PMID 21278728. S2CID 4848298.
- ^ Blaun, Randy; Andreas Wiesenack (1996). "How to Eat Smart". Psychology Today. 29 (3): 34. Retrieved 12 April 2011.
- ^ "The Human Brain-Fats". The Franklin Institute Online. Archived from the original on 26 May 2011. Retrieved 12 April 2011.
- ^ Moser D. J.; Boles Ponto L. L.; Miller I. N.; Schultz S. K.; Menda Y.; Arndt S.; Nopoulos P. C. (2012). "Cerebral blood flow and neuropsychological functioning in elderly vascular disease patients". Journal of Clinical and Experimental Neuropsychology. 34 (2): 220–225. doi:10.1080/13803395.2011.630653. PMC 3582376. PMID 22149630.
- ^ "The Human Brain-Carbohydrates". The Franklin Institute Online. Archived from the original on 7 May 2011. Retrieved 12 April 2011.
- ^ "Brain Foods". Dr. Sears Official Website. Retrieved 12 April 2011.
- ^ Neal E. G.; Chaffe H.; Schwartz R. H.; Lawson M. S.; Edwards N.; Fitzsimmons G.; Cross J. H. (2008). "The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial". The Lancet Neurology. 7 (6): 500–506. doi:10.1016/s1474-4422(08)70092-9. PMID 18456557. S2CID 23670644.
- ^ Henderson S. T. (2008). "Ketone bodies as a therapeutic for Alzheimer's disease". Neurotherapeutics. 5 (3): 470–480. doi:10.1016/j.nurt.2008.05.004. PMC 5084248. PMID 18625458.
- ^ Yeh Y. Y.; Zee P. (1976). "Relation of ketosis to metabolic changes induced by acute medium-chain triglyceride feeding in rats". The Journal of Nutrition. 106 (1): 58–67. doi:10.1093/jn/106.1.58. PMID 1245892.
- ^ Hurtado, Linda (5 June 2013). "Local doctor says coconut oil helps reduce symptoms of Alzheimer's disease".
- ^ "Spring Hill couple inspires research into coconut oil for Alzheimer's patients". 3 June 2013.
- ^ "The Human Brain-Protein". The Franklin Institute Online. Archived from the original on 26 May 2011. Retrieved 13 April 2011.
- ^ Lister J. P., Blatt G. J., DeBassio W. A., Kemper T. L., Tonkiss J., Galler J. R., Rosene D. L.; Blatt; Debassio; Kemper; Tonkiss; Galler; Rosene (2005). "Effect of prenatal protein malnutrition on numbers of neurons in the principal cell layers of the adult rat hippocampal formation". Hippocampus. 15 (3): 393–403. doi:10.1002/hipo.20065. PMID 15669101. S2CID 45527453.CS1 maint: multiple names: authors list (link)
- ^ Reeds, Peter J.; Burrin, Douglas G.; Stoll, Barbara; Jahoor, Farook (2000). "Intestinal Glutamate Metabolism". The Journal of Nutrition. 130 (4S Suppl): 978S–82S. doi:10.1093/jn/130.4.978S. PMID 10736365.
- ^ "Glutamate formiminotransferase deficiency: MedlinePlus Genetics".
- ^ Ankarcrona, Maria; Dypbukt, Jeannette M.; Bonfoco, Emanuela; Zhivotovsky, Boris; Orrenius, Sten; Lipton, Stuart A.; Nicotera, Pierluigi (1995). "Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function". Neuron. 15 (4): 961–73. doi:10.1016/0896-6273(95)90186-8. PMID 7576644. S2CID 14168737.
- ^ Hulsebosch, Claire E.; Hains, Bryan C.; Crown, Eric D.; Carlton, Susan M. (2009). "Mechanisms of chronic central neuropathic pain after spinal cord injury". Brain Research Reviews. 60 (1): 202–13. doi:10.1016/j.brainresrev.2008.12.010. PMC 2796975. PMID 19154757.
- ^ Broadley, KJ (March 2010). "The vascular effects of trace amines and amphetamines". Pharmacology & Therapeutics. 125 (3): 363–75. doi:10.1016/j.pharmthera.2009.11.005. PMID 19948186.
- ^ Pendleton, RG; Gessner, G; Sawyer, J (September 1980). "Studies on lung N-methyltransferases, a pharmacological approach". Naunyn-Schmiedeberg's Archives of Pharmacology. 313 (3): 263–8. doi:10.1007/BF00505743. PMID 7432557. S2CID 1015819.
- ^ Pascucci T., Ventura R., Puglisi-Allegra S., Cabib S.; Ventura; Puglisi-Allegra; Cabib (2002). "Deficits in brain serotonin synthesis in a genetic mouse model of phenylketonuria". NeuroReport. 13 (18): 2561–2564. doi:10.1097/00001756-200212200-00036. PMID 12499868. S2CID 38099620.CS1 maint: multiple names: authors list (link)
- ^ Thompson A. J., Tillotson S., Smith I., Kendall B., Moore S. G., Brenton D. P.; Tillotson; Smith; Kendall; Moore; Brenton (1993). "Brain MRI changes in phenylketonuria: associations with dietary status". Brain. 116 (4): 811–821. doi:10.1093/brain/116.4.811. PMID 8353710.CS1 maint: multiple names: authors list (link)
- Neuroscience
- Nutritional science